1. Introduction
Over three years, the COVID-19 global crisis has offered valuable lessons that can benefit humanity when facing similar disasters in the future [
1]. The outbreak also indirectly highlights the varied responses and approaches adopted by different countries in managing the disease. Resulting proposed actions, such as the implementation of strict measures and financial support, notably proved effective in reducing infection and fatality rates [
2]. Furthermore, a substantial amount of genomic data related to SARS-CoV-2, the virus causing COVID-19, has been systematically stored in popular databases. This wealth of information allowed scientists to extract insights and knowledge for effectively combating the pandemic. Utilizing this dataset, numerous studies have been conducted such as differentiating the DNA sequence of the COVID-19 virus from other diseases with similar symptoms [
3,
4,
5] and classifying these sequences based on virus variants [
6,
7]. By leveraging AI technology, these efforts can provide benefits for cost-effective disease detection.
Initially, no significant mutations were observed in the COVID-19 virus during the first 8 months following the World Health Organization’s declaration of it as a global threat [
8]. However, a series of variants of concern gained prominence shortly thereafter, starting with the Alpha variant that emerged in September 2020 [
9] and concluding with the Omicron variant, which became the predominant variant worldwide as of January 2022 [
10]. Studies indicate that human migration (gene flow) plays a crucial role in virus evolution [
11], allowing for the generation of many variations. The increased occurrence of distinct mutations in European, Asian, and North American sequences suggests the virus’s tendency to cluster geographically [
12]. Moreover, analyzing the variations in the coronavirus genome among 10 countries, including the Czech Republic, France, Thailand, the USA, Japan, Taiwan, China, Australia, Greece, and India, concludes that the structure of the coronavirus genome significantly differs among countries [
13]. In a specific area, a genome-wide analysis of circulating SARS-CoV-2 strains has been conducted to identify the emergence of novel co-existing mutations and trace their geographical distribution within India [
14]. Thus, the genetic variances observed among the strains from diverse locations can be linked to their geographical distributions.
When considering the impact of the COVID-19 outbreak, a comprehensive analysis of the SARS-CoV-2 genome sequences isolated from individuals in six geographic areas reveals correlations with fatality rates in various countries [
15]. This correlation is also supported by studies indicating a positive connection between the occurrence of specific gateway mutations and mortality [
16,
17]. Conversely, some studies suggest no association between SARS-CoV-2 variants and mortality rates [
18,
19,
20]. Nevertheless, the diverse structure of the coronavirus genome among countries is a critical aspect that requires investigation for vaccine development [
13]. In addition to extensive work in phylogenetic-based approaches, which includes a specific profiling method for DNA sequences aimed at the accurate analysis of SARS-CoV-2 genomes at the genus or species levels [
21], it is equally important to explore country-based classifications of sequences. To the best of our knowledge, no existing literature has explicitly conducted a classification of DNA sequences based on the country where they come from. While we cannot ignore the fact that each country has varying land areas that may influence the level of virus variation in each region, recognizing the distinction of DNA sequences, a pivotal component in the genomic data, based on the country of origin could potentially deepen our understanding of how the virus evolves within the area.
As the home to more than 9% of the world’s population and a region with a surge in infection cases since March 2020 [
22], it is crucial to investigate the role of geographical proximity in shaping the genetic structure of the SARS-CoV-2 genome in the Association of Southeast Asian Nations (ASEAN) countries. Several strains that were isolated before the national implementation of border control exhibited a high degree of invariance, while others demonstrated approximately 80% of synonymous mutations, suggesting possible ongoing virus adaptation in the region [
23]. Focused on DNA, research related to sequence profiling has been carried out regionally, involving five ASEAN countries with a relatively limited number of sequences [
24]. To enhance the analysis with a larger dataset and provide clear information on how the virus’ DNA differs between countries, particularly where the countries are geographically close to each other, this paper purposes to classify SARS-CoV-2 DNA sequences based on the country using a deep learning model. Concentrating on nine ASEAN countries, we also primarily explore the connection between the classification results and each country’s international policy restrictions to gain insight into how the virus behaves in different places under different rules.
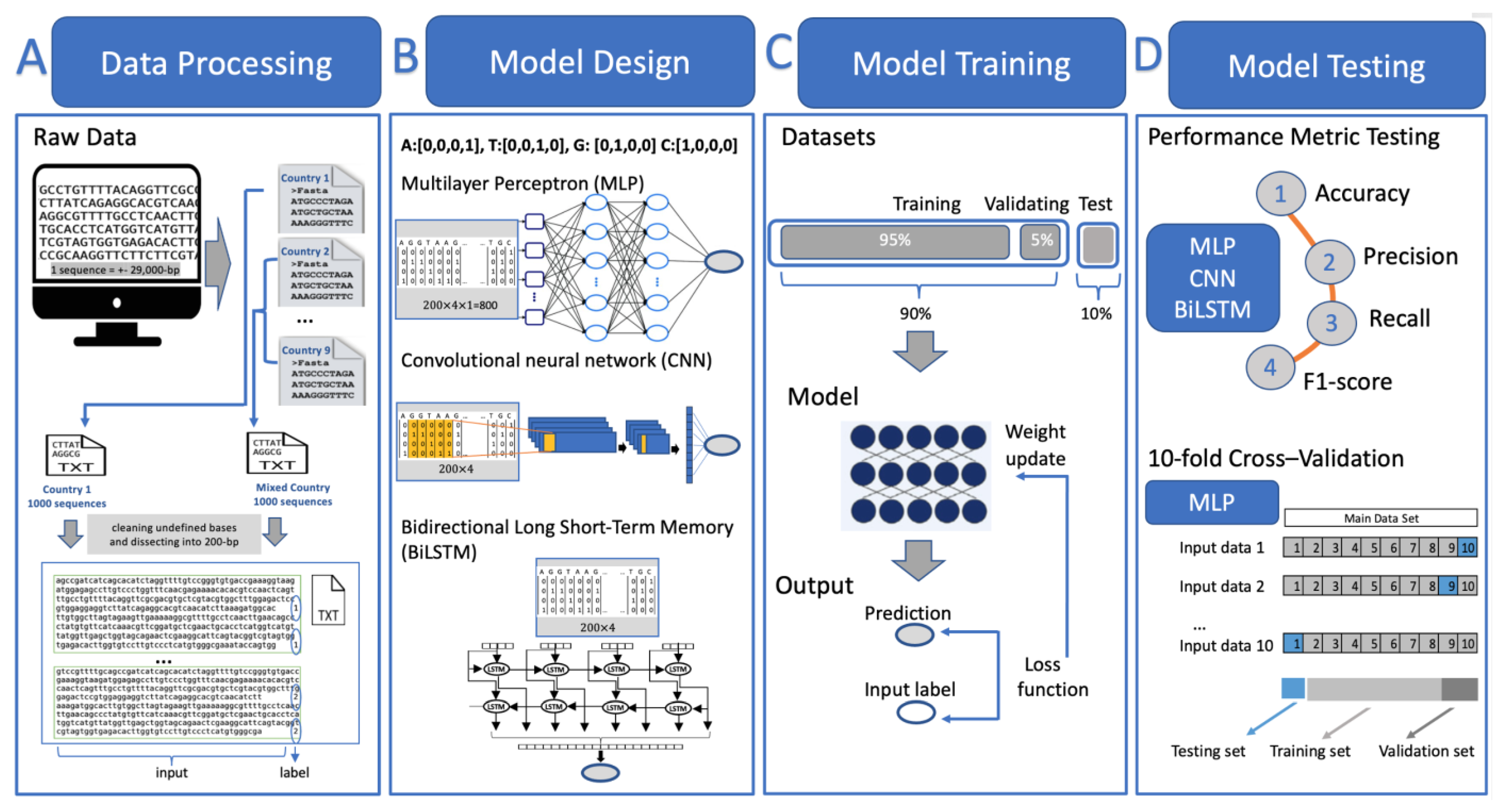
3. Results and Discussion
Figure 4 displays the accuracy chart of DNA sequence classification for the MLP, CNN, and BiLSTM models. When considering all available sequences, as depicted in
Figure 4a, it is shown that sequences from Brunei can be distinguished from those of other ASEAN countries with almost perfect accuracy, reaching 98%. Conversely, the three countries with a lower accuracy are Indonesia, Malaysia, and Vietnam. This pattern remains relatively similar when exclusively involving the Delta variant in the dataset for each country, as shown in
Figure 4b. Therefore, even though the variant is the same, the DNA sequences can still be differentiated based on the country of origin.
Regarding the impact of different areas among ASEAN countries, while Brunei exhibits a higher sequence accuracy than Malaysia, for instance, it is noteworthy to consider other classification results depicted in
Figure 4. In this figure, the sequence accuracy of Singapore, despite being a small country, is not higher than that of Cambodia and Laos, two countries with larger geographical areas. In future work, it would be intriguing to explore the correlation between a country’s size and the diversity of its DNA sequences for generalizing this finding.
Table 4 demonstrates the detailed performance metrics for each model, involving Delta variant sequences. The execution time, which measures the time in seconds at which the code is executed for the dataset of each corresponding country, is also calculated. Notably, from
Table 4, the execution time of the MLP is relatively faster than that of the CNN or BiLSTM, with a comparable level of accuracy. Hence, the MLP model proves to be suitable in terms of both accuracy and execution time. For further assurance in evaluating the performance of the MLP model,
Table 5 shows the classification accuracy resulting from the 10-fold cross-validation technique. It is obvious from
Table 5 that the accuracy results are relatively similar to those of the previous scenario where the model was trained without the implementation of cross-validation.
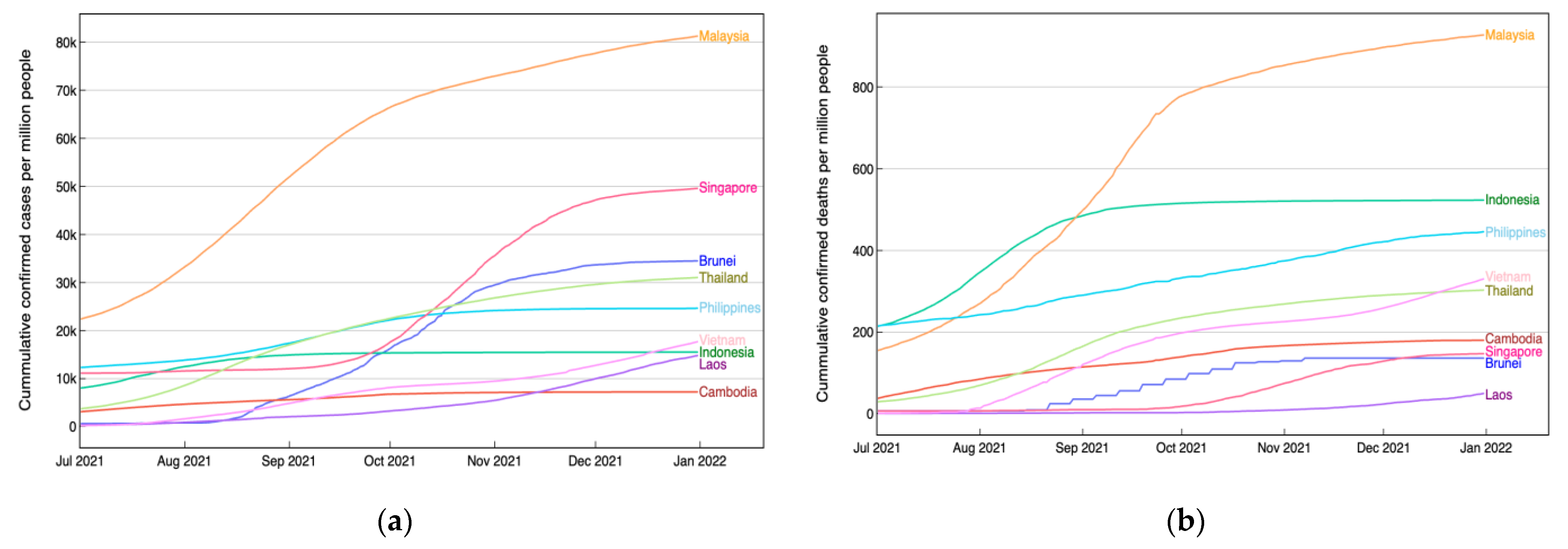
We now analyze these classification results in the context of the pandemic situation and the implementation of international travel policy. Initially, we observed the pandemic situation in each country, focusing particularly on the number of cases and deaths accumulated during the period from July to December 2021. Referring to the second dataset from official data collated by Our World in Data [
26], as depicted in
Figure 5, it appears non-coincidental that Malaysia, one of the countries with a lower accuracy, experienced a severe condition with a notably high number of both accumulated new cases and deaths per million people. In contrast, Cambodia and Laos exhibited relatively lower numbers. While it is premature to conclude that a country with high accuracy, in terms of its DNA sequence, which can be distinguished well from others, tends to undergo a favorable COVID-19 condition with fewer accumulated cases and deaths, we consider this as trigger information to be explored separately in the future.
For additional context, without considering the DNA sequence, a previous study examining pandemic trajectories at the beginning of the outbreak in four ASEAN countries revealed that Malaysia and Vietnam started with strong early performances, while the crisis began severely in the Philippines and Singapore [
28]. However, challenges persisted in Malaysia and the Philippines, with relative stability in Vietnam and Singapore. The study emphasized that significant political uncertainty persisted, exemplified by the fragile political situation in Malaysia in 2021. Given that the effectiveness of planning and executing a comprehensive multisectoral response is heavily dependent on national leadership, initial resources obviously play a crucial role, but policy actions also carry significant weight amid the fight against the pandemic.
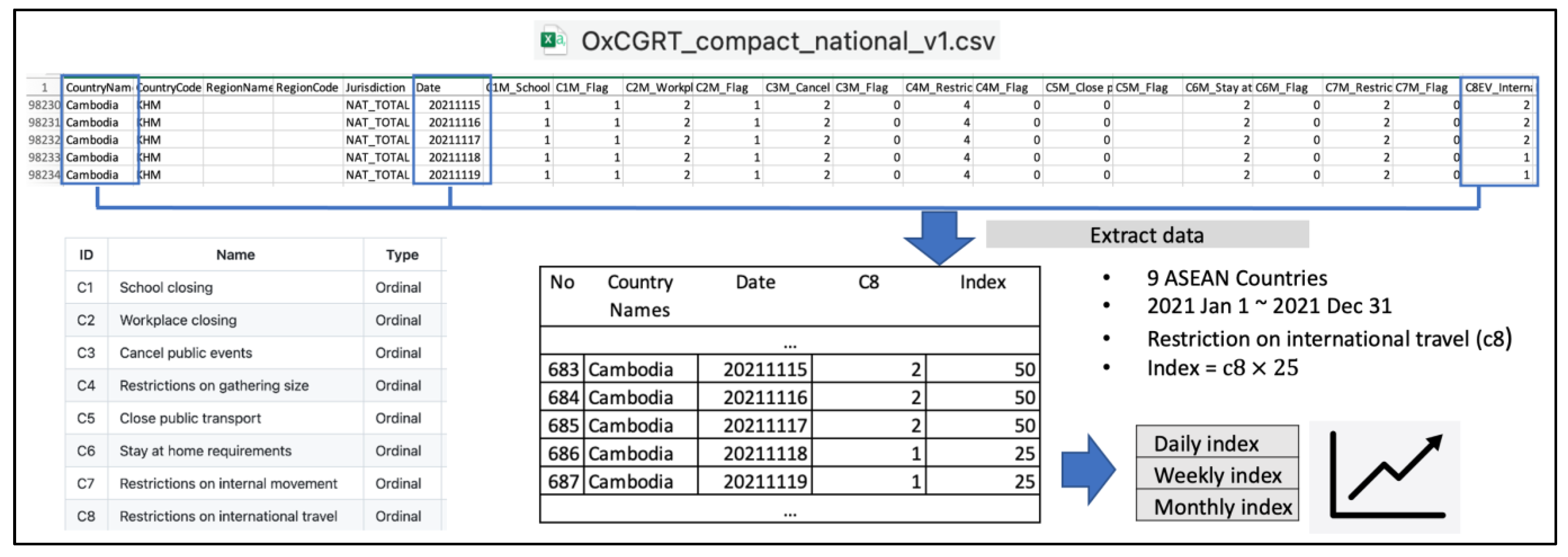
Moving to the main purpose of the study, we further explored the potential correlation between classification accuracy and policy implementation in each country. We utilized the international travel policy index resulting from the processing of the third dataset, accessed from The Oxford COVID-19 Government Response Tracker (OxCGRT) project [
27]. The strict implementation of international travel policy, compared with other government policies, might correlate with the evolution of the virus within a country. Therefore, correlating this policy with classification accuracy provides a preliminary insight into its effectiveness in safeguarding the country from pandemic outbreaks. Before observing the correlation, let us direct our focus to
Figure 6, which provides a graphical representation of the monthly international travel policy index in each country throughout 2021.
Taking a closer look at
Figure 6, we noticed a consistent application of strict international travel policy by Vietnam throughout 2021, while other countries, particularly Laos, exhibited frequent changes in their restriction scale almost every month. The majority of lines overlap at an index of 75, indicating that most countries implemented bans on arrivals from some regions. Vietnam stands out as the country that implemented a total border closure for more than half of the year, while Singapore noticeably eased restrictions to only screening arrivals in the last four months of 2021. Above all, none of the ASEAN countries had completely open borders during the 2021 timeframe.
To investigate whether the international travel policy index correlates with the accuracy of DNA sequence classification within each country, we determined the correlation coefficient between the policy index in a specific month and the classification accuracy in a subsequent month in the second half of 2021. We only considered six countries, excluding Brunei, Laos, and the Philippines due to insufficient monthly datasets. Referring to the emergence of the SARS-CoV-2 Omicron variant [
29], where a new wave of infection is expected approximately every additional (up to) 4 months of virus circulation (although we cannot confirm the periodicity will be maintained), we calculated the correlation of the policy index implemented up to 4 months before the collection date of DNA sequences. For an initial observation,
Figure 7 illustrates the chart of the monthly policy index implemented up to 4 months before the sequence collection date, alongside the monthly classification accuracy for each country. It is important to note that for this month-wise classification, we conducted a similar classification strategy, but the involved sequence was reduced to 200 sequences for each class with a batch size of 100.
To indicate a potential link between prior
i-month policy indices and the accuracy of DNA sequences in the corresponding month,
Table 6 shows diverse correlation coefficients for each country. To address the issue of a very small sample size [
30], we calculated Pearson correlations between the weekly policy index and the accuracy of week-wise classification, with 25 sequences per class, in a six-months dataset. This calculation means that each entry in
Table 6 results from the Pearson correlation and
p-value calculation involving 24 pairs of data. Except for Indonesia, the majority of correlations demonstrated a positive trend, suggesting that, if a relationship exists, the policy index positively correlates with the classification accuracy.
Utilizing the p-value with a significance level of 5%, we observed a significant relationship between classification accuracy in Vietnam and the policy implemented four months before the collection date of the DNA sequence. Here, the p-value equals 0.01, which is less than 5%, indicating that the correlation is statistically significant with a coefficient of 0.49. This suggests that the international travel policy in March positively corelates with classification accuracy in July, while the policy in April corresponds to accuracy in August, and so forth. However, with the same significance level, the correlation between the accuracy and policy index of the preceding 3 months is the highest in Vietnam, standing at 0.77. Therefore, the classification accuracy in Vietnam is highly positively correlated with the policy index three months before the collection date of DNA sequences. Similar to the interpretation for Vietnam, the accuracy in Malaysia exhibits a positive correlation with the policy index one month before the DNA sequence collection date, with a moderate relationship standing at 0.55. Meanwhile, the accuracy in Indonesia is negatively correlated with the policy index up to four months before the DNA sequence collection date.
Unfortunately, at a 5% significance level, it was determined that there is no significant correlation observed between the policy index and DNA sequence classification accuracy for other countries. However, it is crucial to approach the interpretation of data cautiously and not consider a
p-value of 0.05 as a definitive threshold [
31]. Relying on the
p-value derived from a sole statistical test to assess the scientific validity of research constitutes an inappropriate use of the
p-value. Nevertheless, it is important to note that alternative approaches to the
p-value also share comparable limitations [
32]. If we are allowed to utilize a significance level set at 15%, similar to Vietnam, DNA sequence classification accuracy in Singapore positively correlates with the policy index applied four months before the sequence collection date. Moreover, classification accuracy in Cambodia and Thailand also positively correlates with its policy implemented in the same month as the collection date, with fair coefficients of 0.31 and 0.34, respectively. While the data show a correlation between the classification accuracy and international travel policy index, it is essential to understand that correlation does not imply causation [
30]. Further investigation is required to explore whether the policy is a causative factor for country-based DNA sequence classification accuracy.
Regarding the relationship between the international travel policy index and the accuracy of DNA sequence classification among ASEAN countries, we found it challenging to definitively conclude that a higher policy index in one country correlated with a higher accuracy in distinguishing its sequences from others. This observation is apparent in
Figure 7, where Singapore consistently maintains a higher accuracy than Vietnam almost over the entire timeframe, despite having a consistently lower policy index every 4 months preceding sequence collection. In addition, Cambodia, with the highest sequence accuracy throughout the period, does not necessarily exhibit the highest policy index among the focused countries. This suggests the presence of additional influencing factors on the accuracy of DNA sequences between each country. For future research, exploring other aspects that may explain why efforts to boost the international travel policy index in each country may not uniformly correlate with results regarding how DNA sequences significantly differ from those of other countries would be intriguing.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}