
A Cosine-Similarity-Based Deconvolution Method for Analyzing Data-Independent Acquisition Mass Spectrometry Data

Abstract
:1. Introduction
- To address the issue of a large amount of data in chromatogram similarity comparison, which create a larger search space and require more calculations. In contrast to the prior method, the MS/MS spectra are at the center of our algorithm, and the information “isolation window” is added when extracting mass spectra data to reduce the comparison space, improving the accuracy of the experiment.
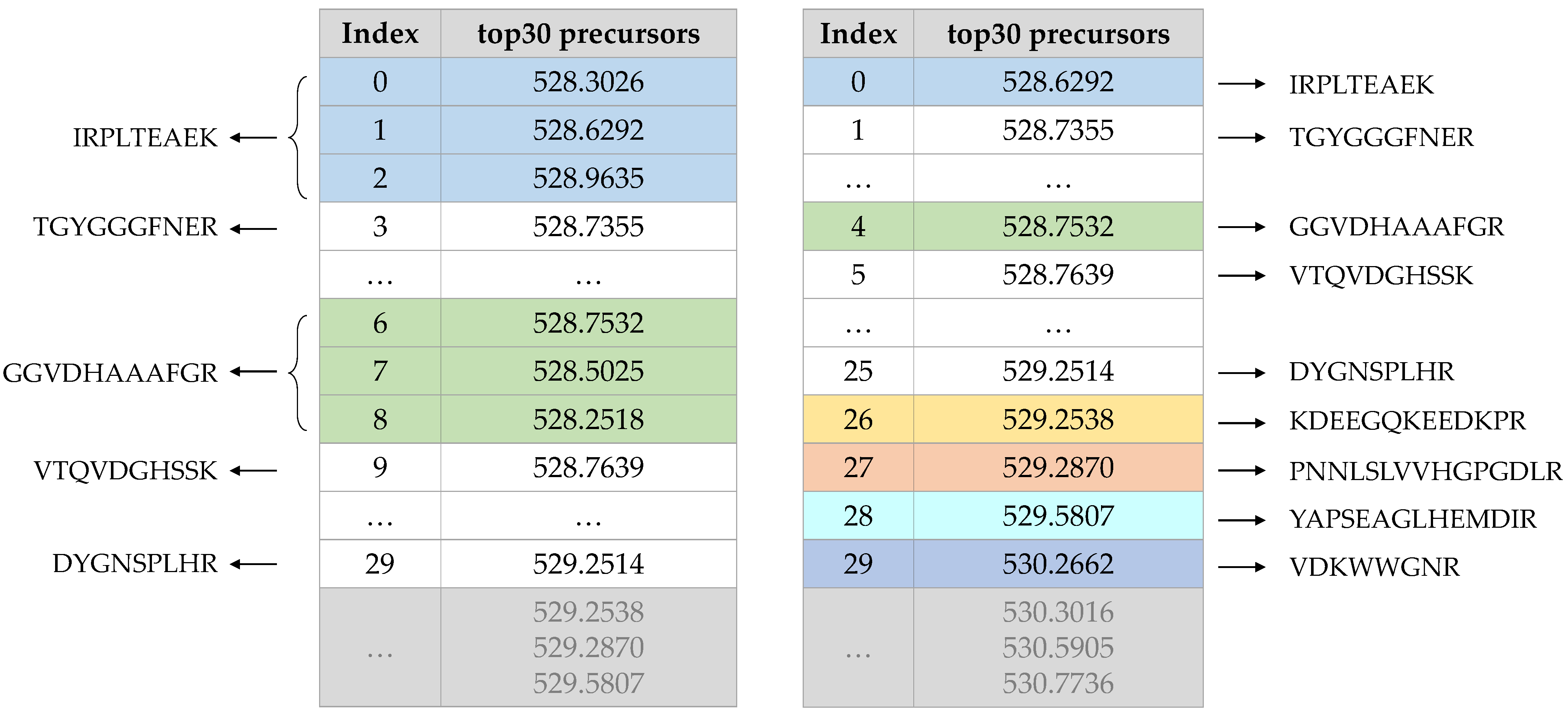
- According to the characteristics of DIA data, there are a large number of isotopic peak clusters in the spectrum. We removed isotopic peak clusters from each MS1 spectrum and carried out an overall search to remove redundancy in each selection of candidate precursors. Reducing the repetition rate ensured that most of the pseudo-MS/MS spectra came from the same peptide.
2. Materials and Methods
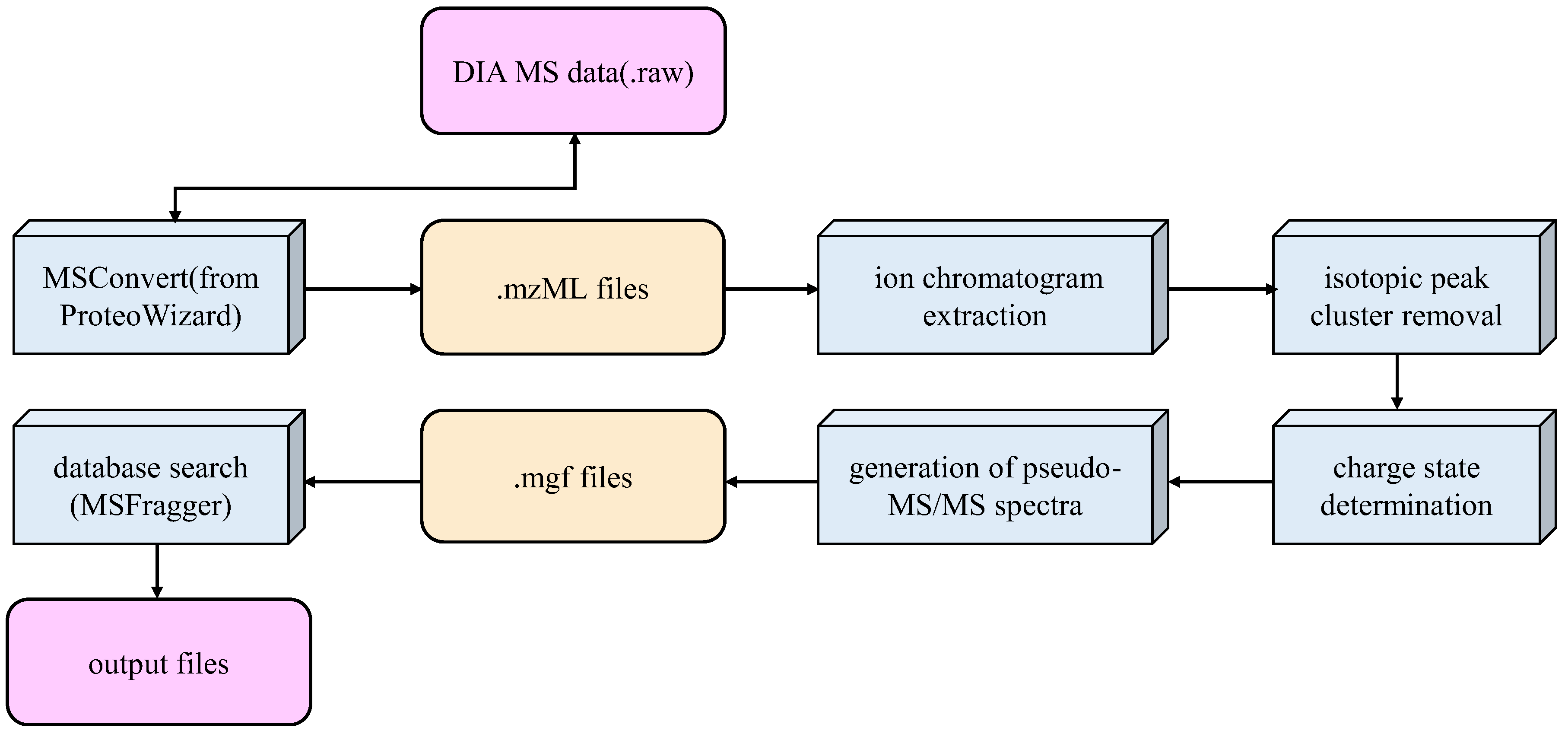
2.1. Raw Mass Spectrometry Data
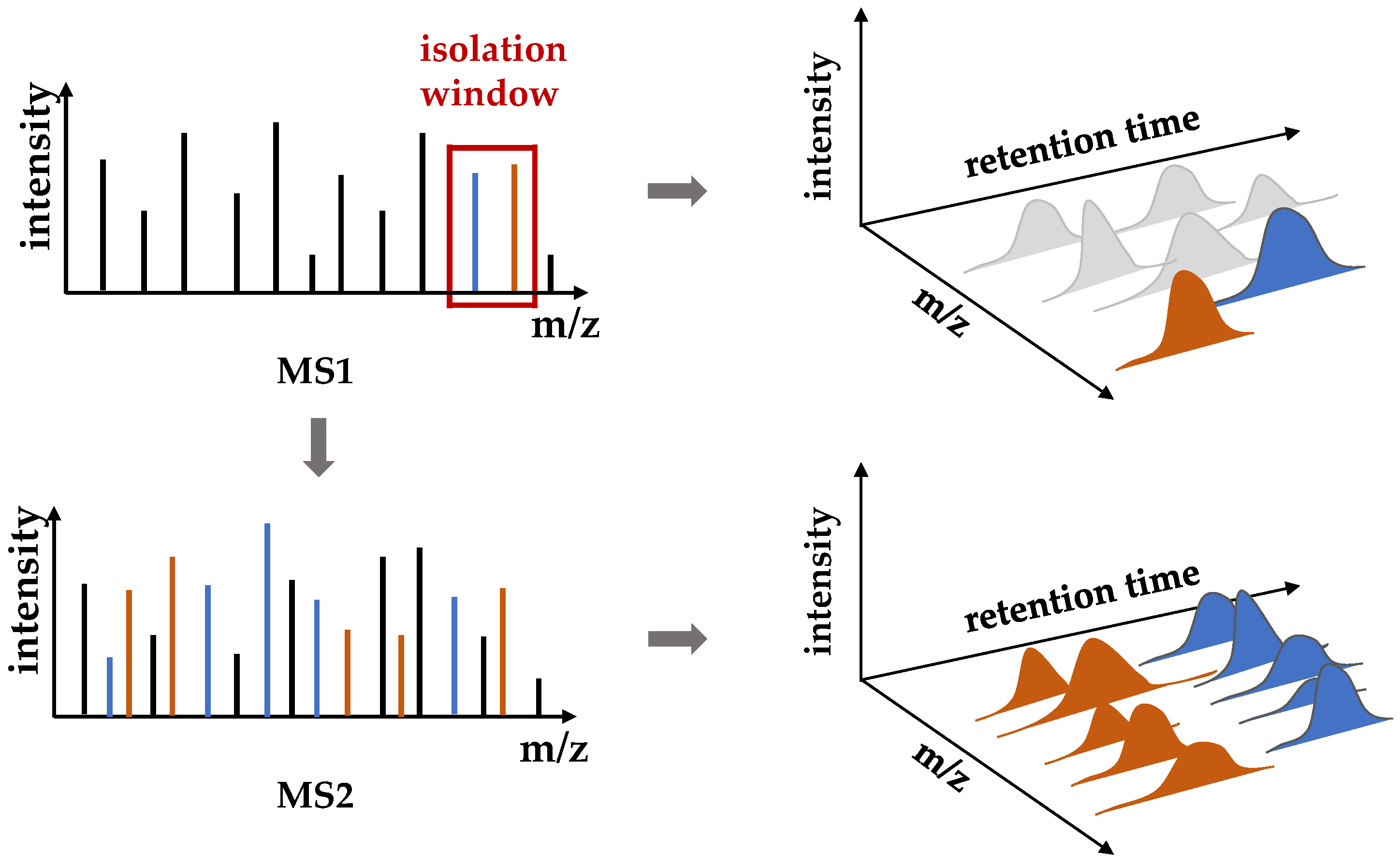
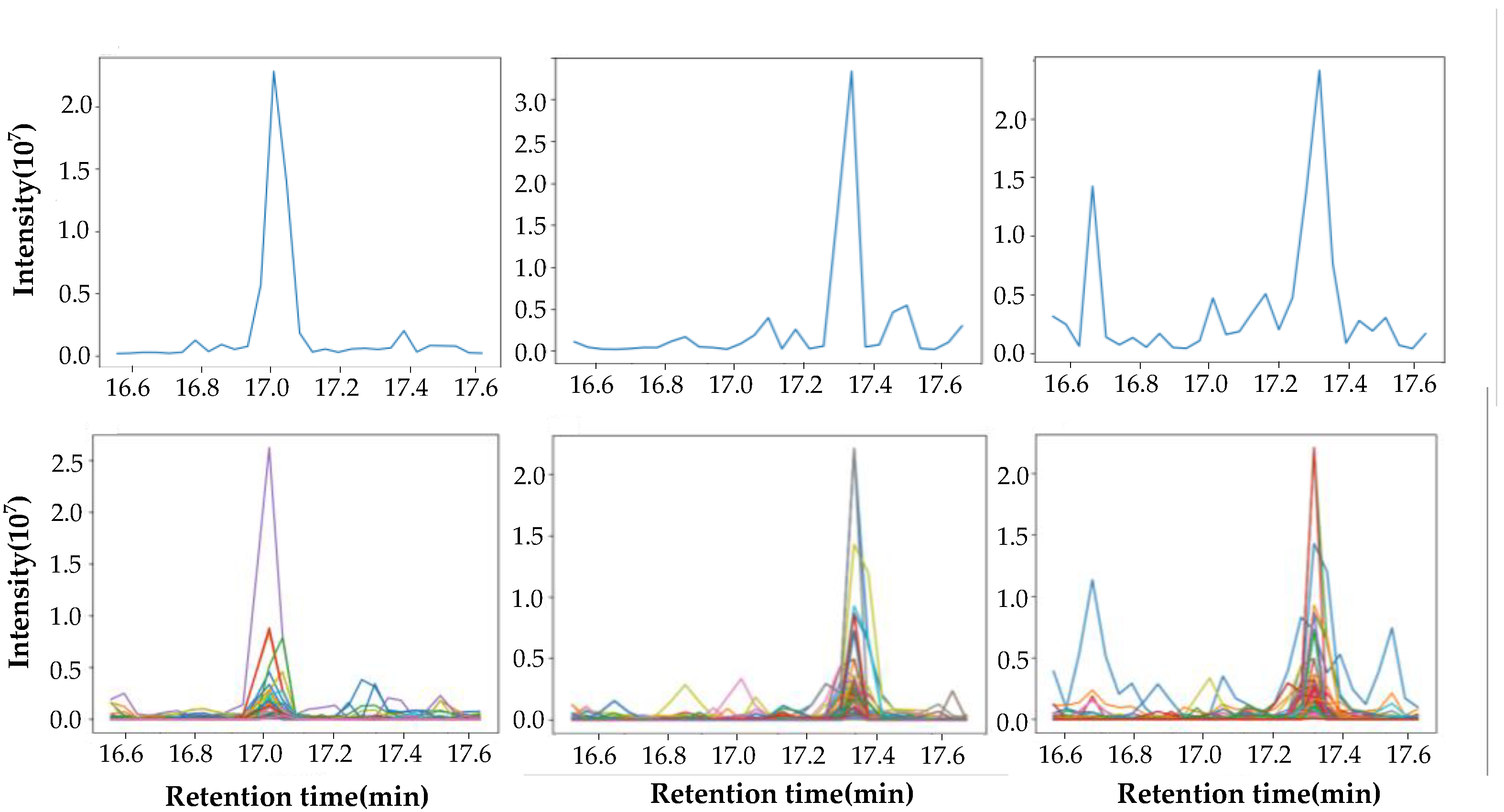
2.2. Data Extraction
2.3. Similarity Comparison
| Algorithm 1: Calculating cosine_similarity of chromatograms of precursors and fragments. |
| Input: Peak intensity list of each precursor item1, peak intensity list of each fragment item2 Output: Cosine_similarity two-dimensional matrix result 1 device ← torch.device (‘cuda’ if torch.cuda.is_available () else ‘cpu’) 2 for i, pre in enumerate (item1) do 3 for j, frg in enumerate (itme2) do 4 x, y ← torch.tensor (np.array([pre, frg]), dtype = torch.float64, device = device) 5 cos ← torch.nn.CosineSimilarity (dim = 0) 6 similar ← cos (x, y) 7 tmp_list ← similar.cpu () 8 result[i] ← tmp_list 9 return result |
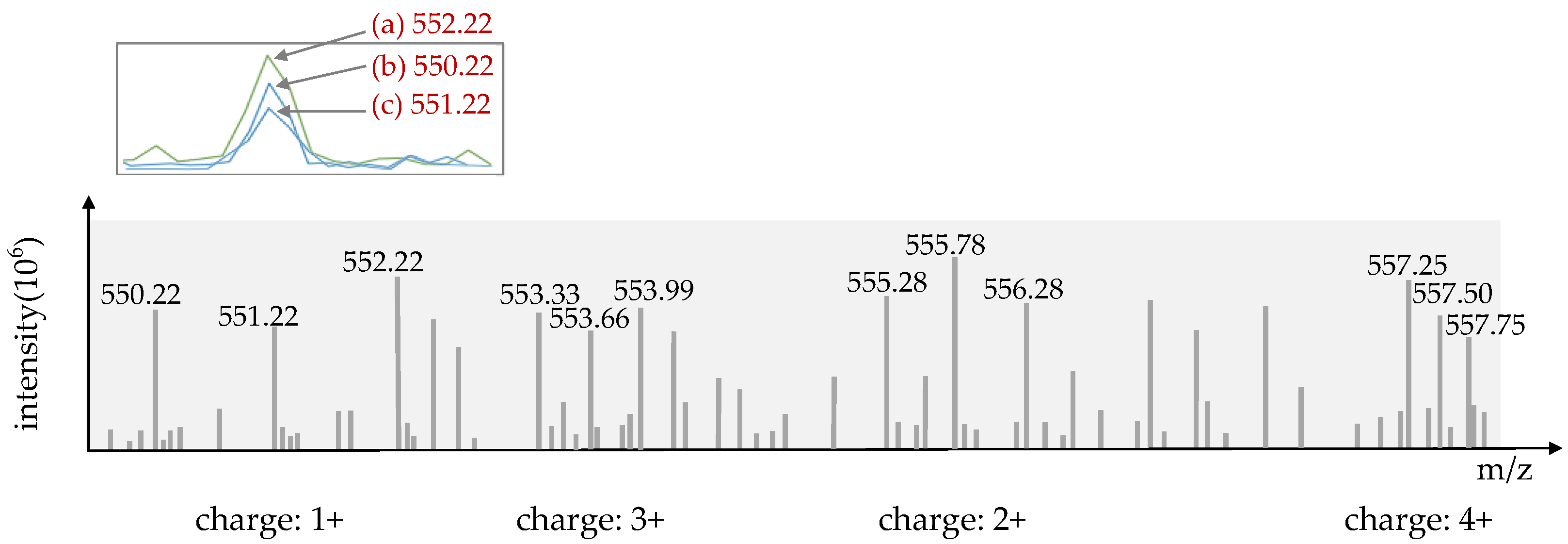
2.4. Remove the Isotopic Peak Cluster and Determine a Precursor’s Charge State
- Confirm mass spectrum scan of the precursor and record its position I in the precursor ion mass spectrum. Each peak in the precursor ion mass spectrum is marked by 0 or 1 to record whether it has been visited;
- Go through all peaks in the precursor ion mass spectrum to find the peak P0 with the highest intensity and record its position I0;
- From I0, move left or right to determine whether the peak difference in mass-to-charge meets the set threshold of mass spectra peaks with P0 (+1: 1.003 da, +2: 0.5015 da, +3: 0.3343 da, +4: 0.2507 da);
- If satisfied, these peaks are then recorded as isotopic clusters of P0, and their charge is confirmed by the threshold. If not, pass the peak;
- Delete the P0 isotopic clusters found above in the original precursor ion mass spectrum so that only the peak with the highest peak intensity is retained; then, find the peak P1 with the highest intensity in all the unvisited peaks and record its position I1;
- Repeat the previous step until all peaks in the precursor ion mass spectrum are visited.
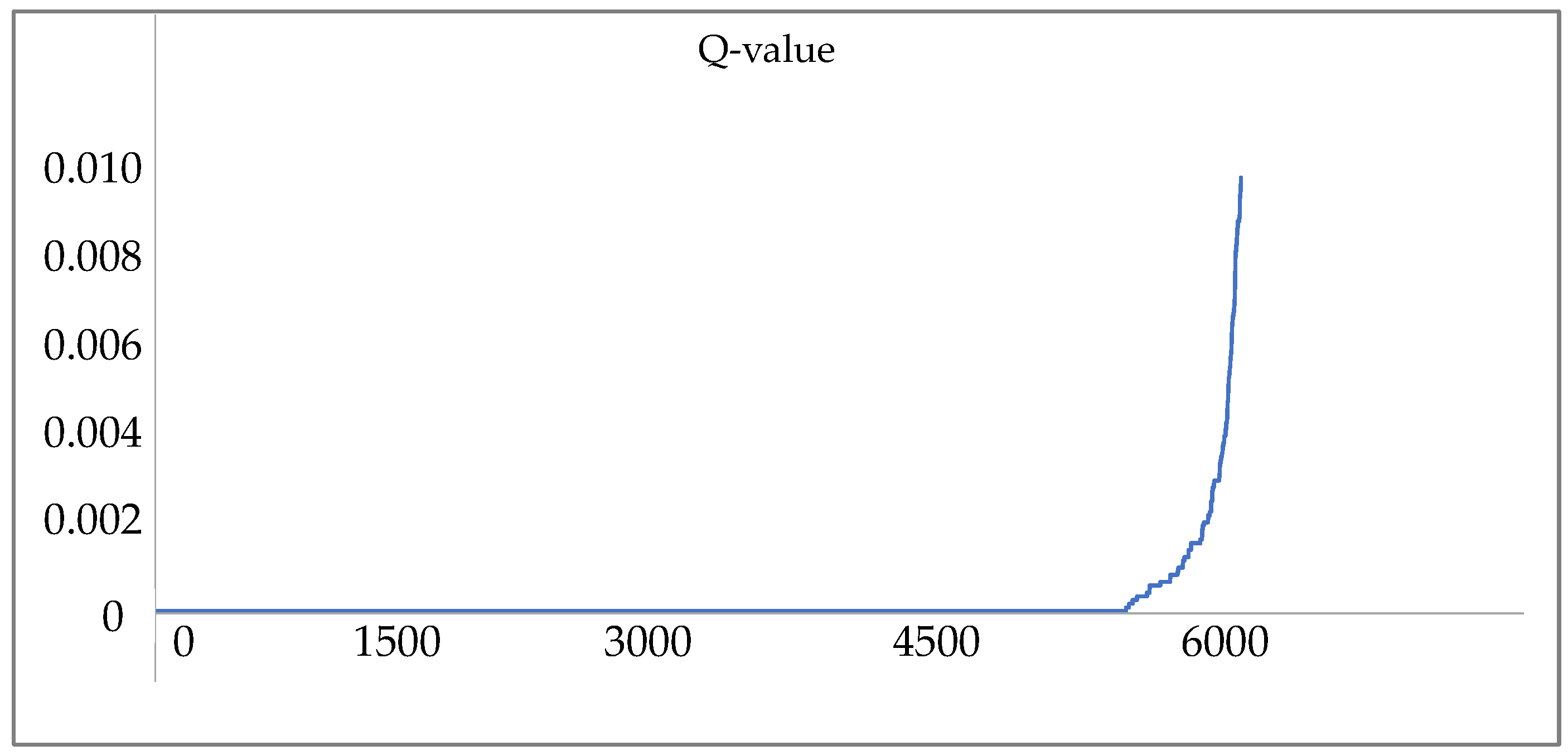
2.5. Peptide and Protein Identification
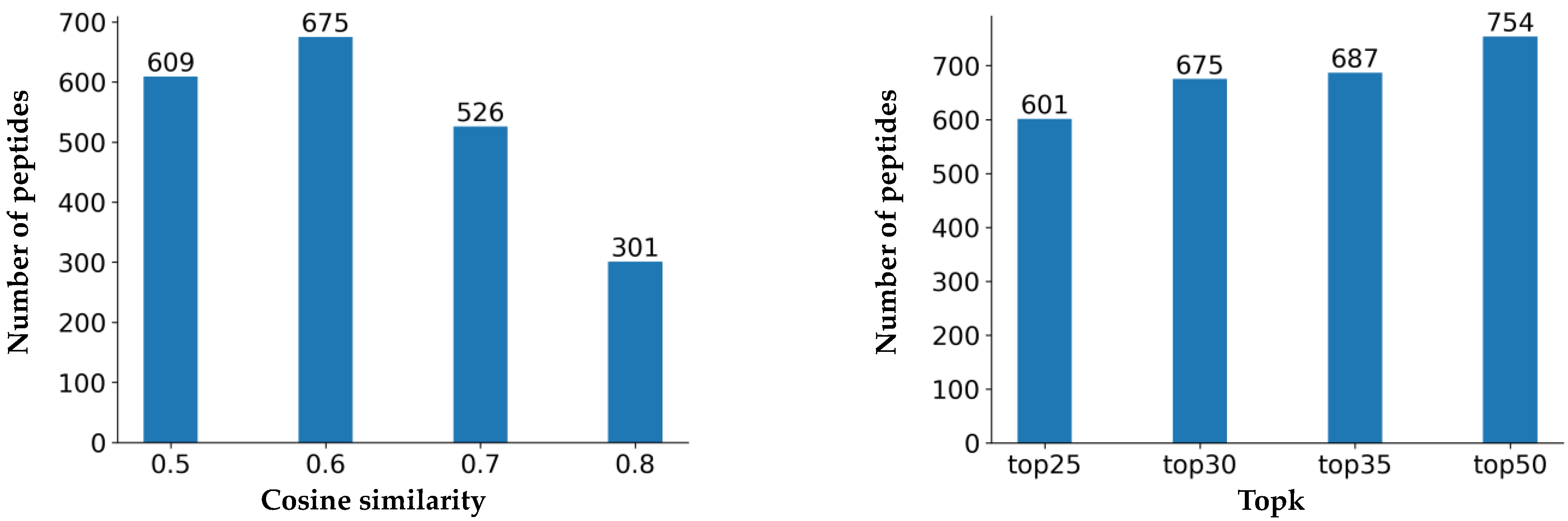
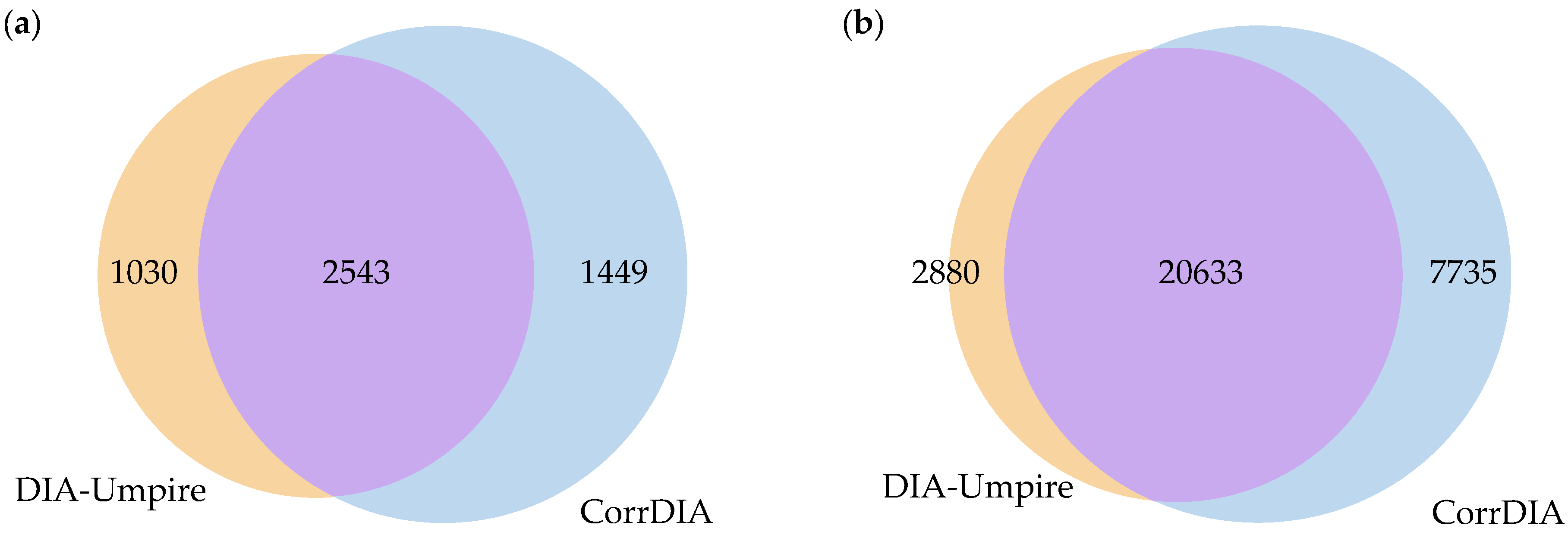
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Software Availability
References
- Steen, H.; Mann, M. The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.S. Protein identification using MS/MS data. J. Proteom. 2011, 74, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Venable, J.D.; Dong, M.Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1, 39–45. [Google Scholar] [CrossRef]
- Röst, H.L.; Rosenberger, G.; Navarro, P.; Gillet, L.; Miladinović, S.M.; Schubert, O.T.; Wolski, W.; Collins, B.C.; Malmström, J.; Malmström, L.; et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014, 32, 219–223. [Google Scholar] [CrossRef]
- Tran, N.H.; Qiao, R.; Xin, L.; Liu, C.Y.; Zhang, X.L.; Shan, B.Z.; Ghodsi, A.; Li, M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 2019, 16, 63–66. [Google Scholar] [CrossRef]
- Bilbao, A.; Varesio, E.; Luban, J.; Strambio-De-Castillia, C.; Hopfgartner, G.; Müller, M.; Lisacek, F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 2015, 15, 964–980. [Google Scholar] [CrossRef]
- Bern, M.; Finney, G.; Hoopmann, M.R.; Merrihew, G.; Toth, M.J.; MacCoss, M.J. Deconvolution of mixture spectra from ion-trap data-independent-acquisition tandem mass spectrometry. Anal. Chem. 2010, 82, 833–841. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zhong, C.Q.; Xu, X.Z.; Cai, S.W.; Wu, X.R.; Zhang, Y.Y.; Chen, J.N.; Shi, J.H.; Lin, S.C.; Han, J.H. Group-DIA: Analyzing multiple data-independent acquisition mass spectrometry data files. Nat. Methods 2015, 12, 1105–1106. [Google Scholar] [CrossRef]
- Peckner, R.; Myers, S.A.; Jacome, A.S.V.; Egertson, J.D.; Abelin, J.G.; MacCoss, M.J.; Carr, S.A.; Jaffe, J.D. Specter: Linear deconvolution for targeted analysis of data-independent acquisition mass spectrometry proteomics. Nat. Methods 2018, 15, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Tada, I.; Chaleckis, R.; Tsugawa, H.; Meister, I.; Zhang, P.; Lazarinis, N.; Dahlén, B.; Wheelock, C.E.; Arita, M. Correlation-based deconvolution (CorrDec) to generate high-quality MS/MS spectra from data-independent acquisition in multisample studies. Anal. Chem. 2020, 92, 11310–11317. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Storey, J.D.; Maccoss, M.J.; Noble, W.S. Posterior error probabilities and false discovery rates: Two sides of the same coin. J. Proteome Res. 2008, 7, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.S.; Johnson, K.A.; Rainville, P.; Smith, B.W.; Wilson, I.D.; Castro-Perez, J.M.; Nicholson, J.K. UPLC/MS(E); a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Spectrom. 2006, 20, 1989–1994. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Bloomfield, N.; Yu, J.S.; White, M.; Kreidl, M.; Egger, A.S.; Freiwald, A.; Ivosev, G.; Wasim, F.; et al. Ultra-fast proteomics with Scanning SWATH. Nat. Biotechnol. 2021, 39, 846–854. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef]
- Chi, H.; Liu, C.; Yang, H.; Zeng, W.F.; Wu, L.; Zhou, W.J.; Niu, X.N.; Ding, Y.H.; Zhang, Y.; Wang, R.M.; et al. Open-pFind enables precise, comprehensive and rapid peptide identification in shotgun proteomics. Nat. Biotechnol. 2018, 36, 1059–1066. [Google Scholar] [CrossRef]
- Shao, G.; Cao, Y.; Chen, Z.L.; Liu, C.; Li, S.T.; Dong, M.Q. How to use open-pFind in deep proteomics data analysis?—A protocol for rigorous identification and quantitation of peptides and proteins from mass spectrometry data. Biophys. Rep. 2021, 7, 207–226. [Google Scholar]
- Yu, F.C.; Teo, G.C.; Kong, A.T.; Li, G.X.; Demichev, V.; Nesvizhskii, A.I. One-stop analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Biotechnol. 2022. preprint. [Google Scholar] [CrossRef]
- Hou, X.H.; Zhou, P.Y.; Gong, P.Y.; Fu, J.L.; Liu, C.; Wang, H.P. Progress in data analysis methods for proteome mass spectrometry based on data-independent acquisition. Prog. Biochem. Biophys. 2022, 49, 2364–2386. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F1 | F2 | F3 | … | Fm – 1 | Fm | |
|---|---|---|---|---|---|---|
| P1 | corr(P1, F1) | corr(P1, F2) | corr(P1, F3) | … | corr(P1, Fm – 1) | corr(P1, Fm) |
| P2 | corr(P2, F1) | corr(P2, F2) | corr(P2, F3) | … | corr(P2, Fm – 1) | corr(P2, Fm) |
| … | … | … | … | … | … | … |
| Pn – 1 | corr(Pn – 1, F1) | corr(Pn – 1, F2) | corr(Pn – 1, F3) | … | corr(Pn – 1, Fm – 1) | corr(Pn – 1, Fm) |
| Pn | corr(Pn, F1) | corr(Pn, F2) | corr(Pn, F3) | … | corr(Pn, Fm – 1) | corr(Pn, Fm) |
| Parameter | Value |
|---|---|
| Precursor tolerance | ±20 ppm |
| Fragment tolerance | ±20 ppm |
| Max variable mods | 3 |
| Peptide length | 5–50 |
| Peptide mass range | 100–5000 |
| Filter | 1% |
| Method | Spectra | Peptide | Proportion |
|---|---|---|---|
| CorrDIA | 34,989 | 28,368 | 81% |
| DIA-Umpire | 33,993 | 23,513 | 69% |
| Sequence | Theoretical m/z | CorrDIA m/z | DIA-Umpire m/z |
|---|---|---|---|
| ALVSGKPAESSAVAATEK | 572.64 | 571.93 | 571.63 |
| QLQQAQAAGAEQEVEK | 864.43 | 864.42 | 863.42 |
| VAVEEVDEEGK | 602.29 | 601.79 | 601.28 |
| LDASESLR | 445.73 | 445.23 | 444.73 |
| LYKEELEQTYHAK | 551.28 | 550.61 | 550.28 |
| Sequence | Theoretical m/z | CorrDIA m/z | Charge | Q-Value |
|---|---|---|---|---|
| VKATNTQHAVEAIR | 513.29 | 512.92 | 3 | 0 |
| WQYGDSAVGR | 380.18 | 379.51 | 3 | 0 |
| GHYTEGAELVDSVLDVVR | 979.99 | 979.99 | 2 | 0 |
| EALEAYR | 426.21 | 425.71 | 2 | 0 |
| HIADLAGNSEVILPVPAFNVINGGSHAGNK | 753.65 | 752.89 | 4 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wu, R.; Qu, Z. A Cosine-Similarity-Based Deconvolution Method for Analyzing Data-Independent Acquisition Mass Spectrometry Data. Appl. Sci. 2023, 13, 5969. https://doi.org/10.3390/app13105969
Zhang X, Wu R, Qu Z. A Cosine-Similarity-Based Deconvolution Method for Analyzing Data-Independent Acquisition Mass Spectrometry Data. Applied Sciences. 2023; 13(10):5969. https://doi.org/10.3390/app13105969
Chicago/Turabian StyleZhang, Xiang, Ruitao Wu, and Zhijian Qu. 2023. "A Cosine-Similarity-Based Deconvolution Method for Analyzing Data-Independent Acquisition Mass Spectrometry Data" Applied Sciences 13, no. 10: 5969. https://doi.org/10.3390/app13105969