
Comprehensive Analysis of the Expression and Clinical Significance of RAS Family Members in Non-Small Cell Lung Cancer Based on Bioinformatics Data and the A549 Cell Line Model

Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Bioinformatics Analysis
2.1.1. TNMplot
2.1.2. The UALCAN Database
2.2. Cell Culture and In Vitro Assay
2.2.1. Material
2.2.2. Cell Culture
2.2.3. Evaluation of Cell Viability with the MTT Test
2.3. Assessment of HRAS and KRAS Gene Expression after Exposure to Deltarasin, Kobe0065 or FTS in A549 Cells
2.3.1. RNA Isolation Exposure of Cells to Tested of Selected Compounds from A549 Cells
2.3.2. Reverse Transcription Reaction
2.3.3. Real Time PCR
2.3.4. Statistical Analysis
3. Results
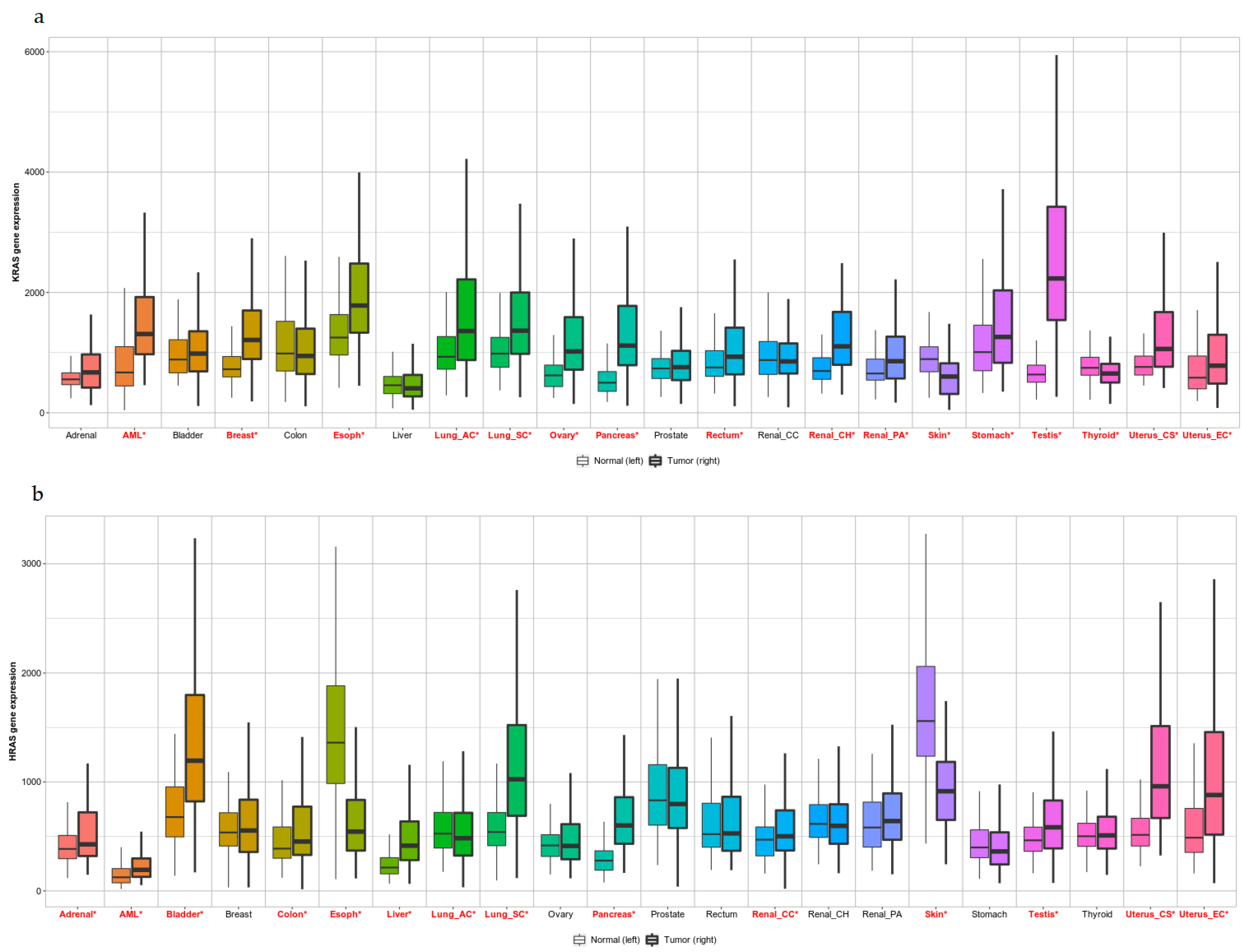
3.1. Pan-Cancer View of KRAS and HRAS Expression Levels
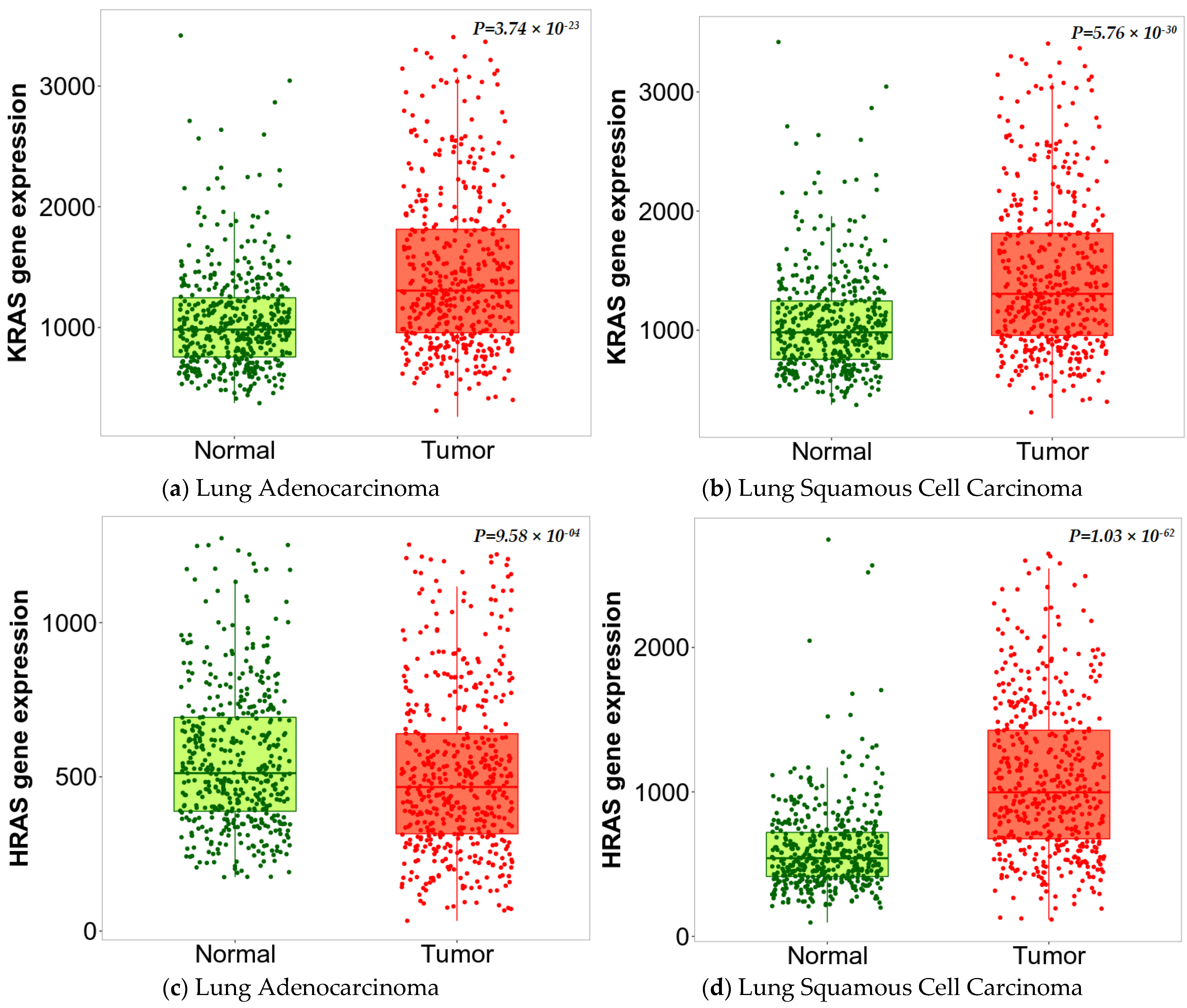
3.2. Expression of KRAS and HRAS Genes in NSCLC
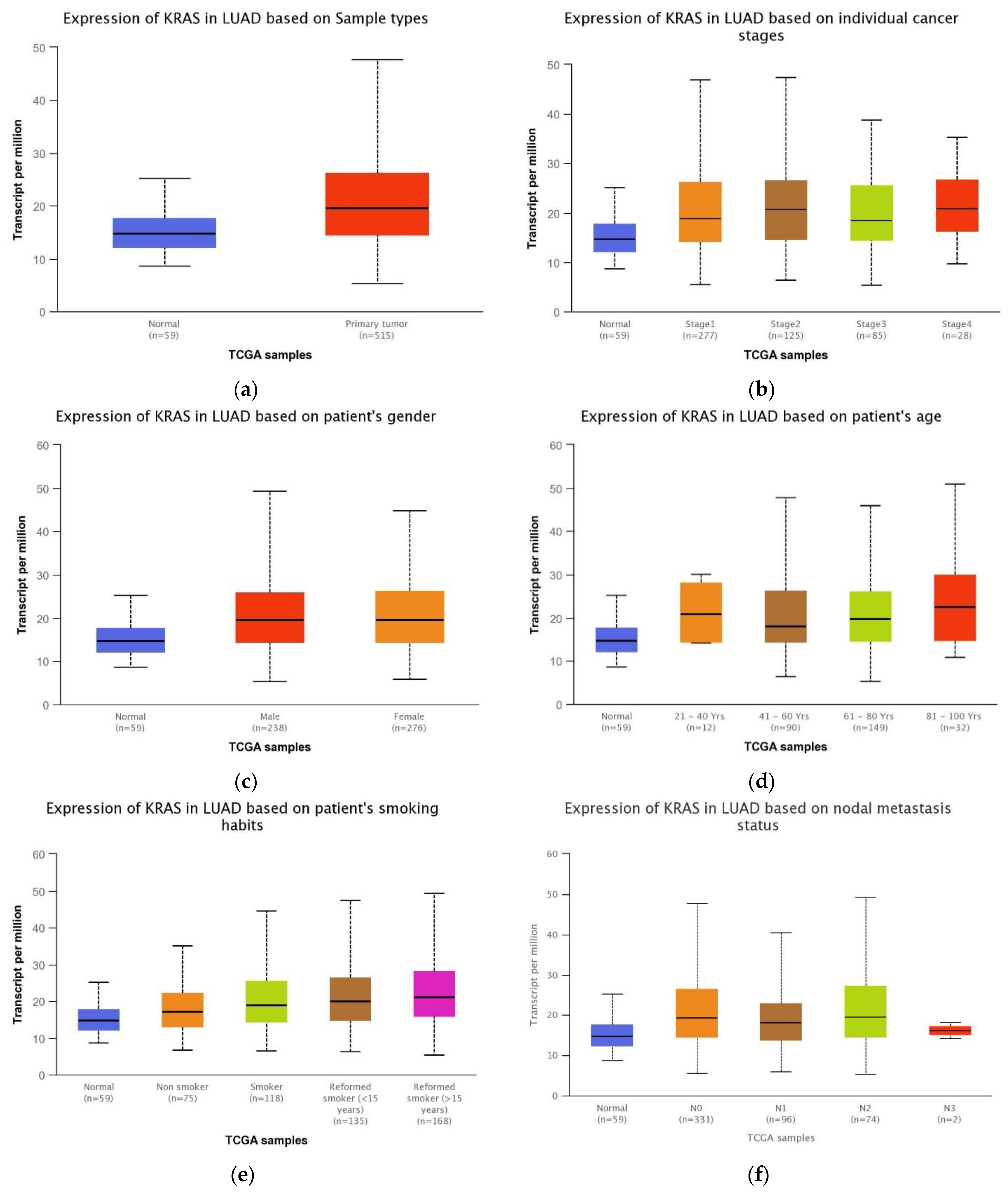
3.3. Association between the KRAS Expression and LUAD Patient Clinical Features
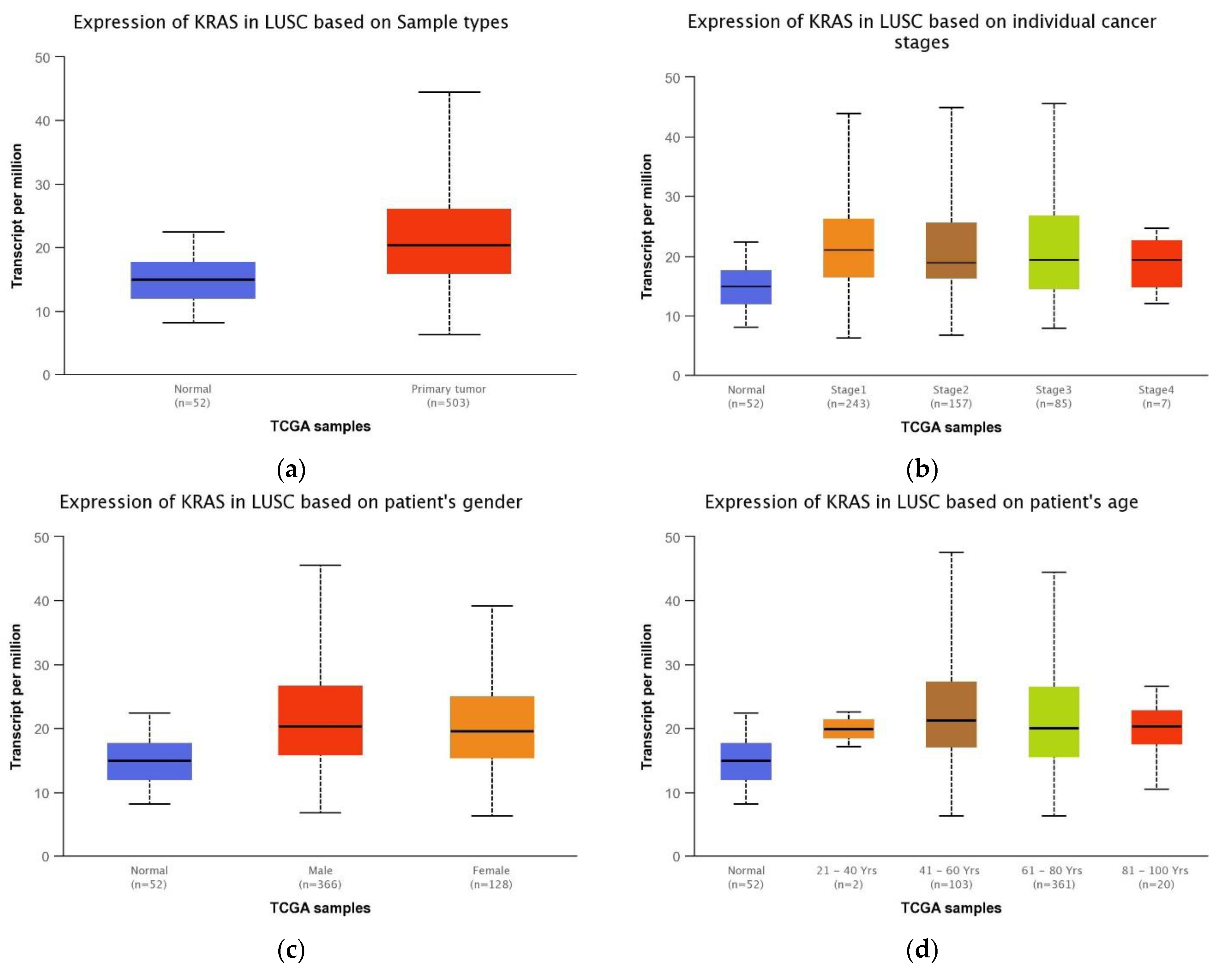
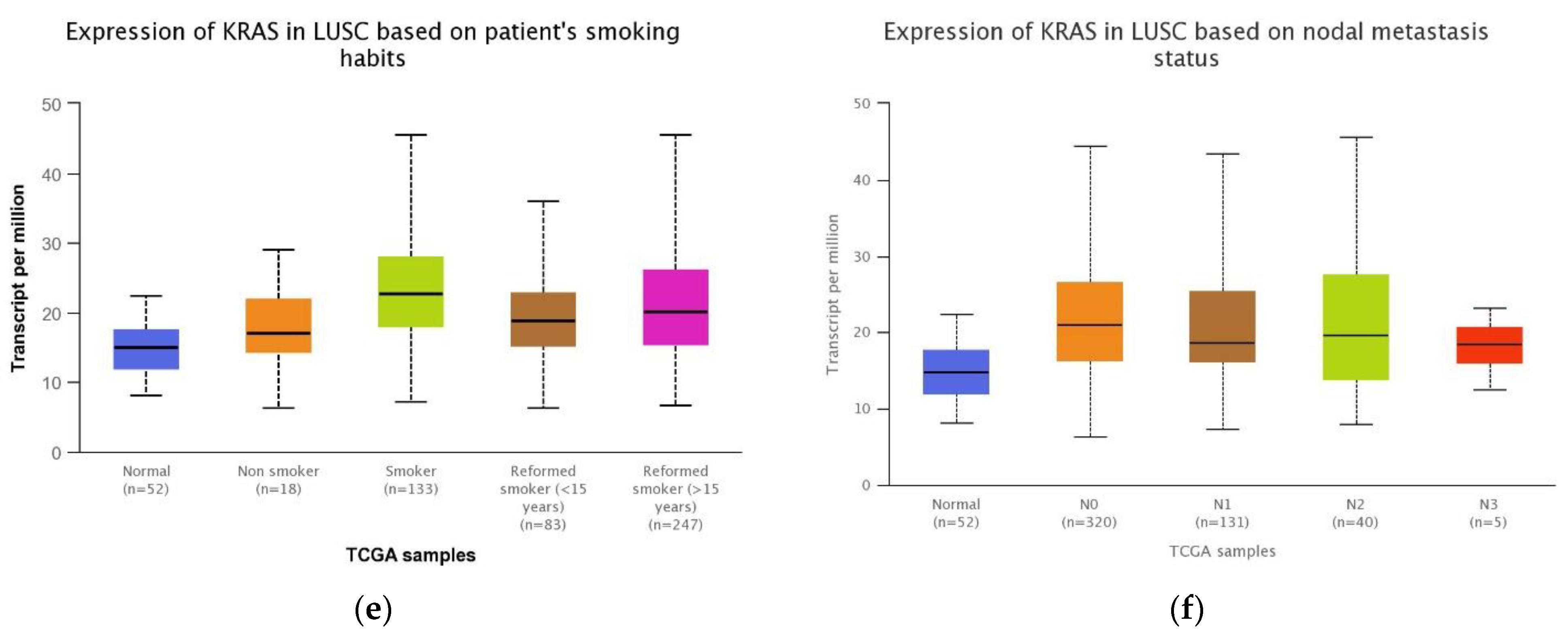
3.4. Association between the KRAS Gene Expression and Clinical Features in LUSC Patients
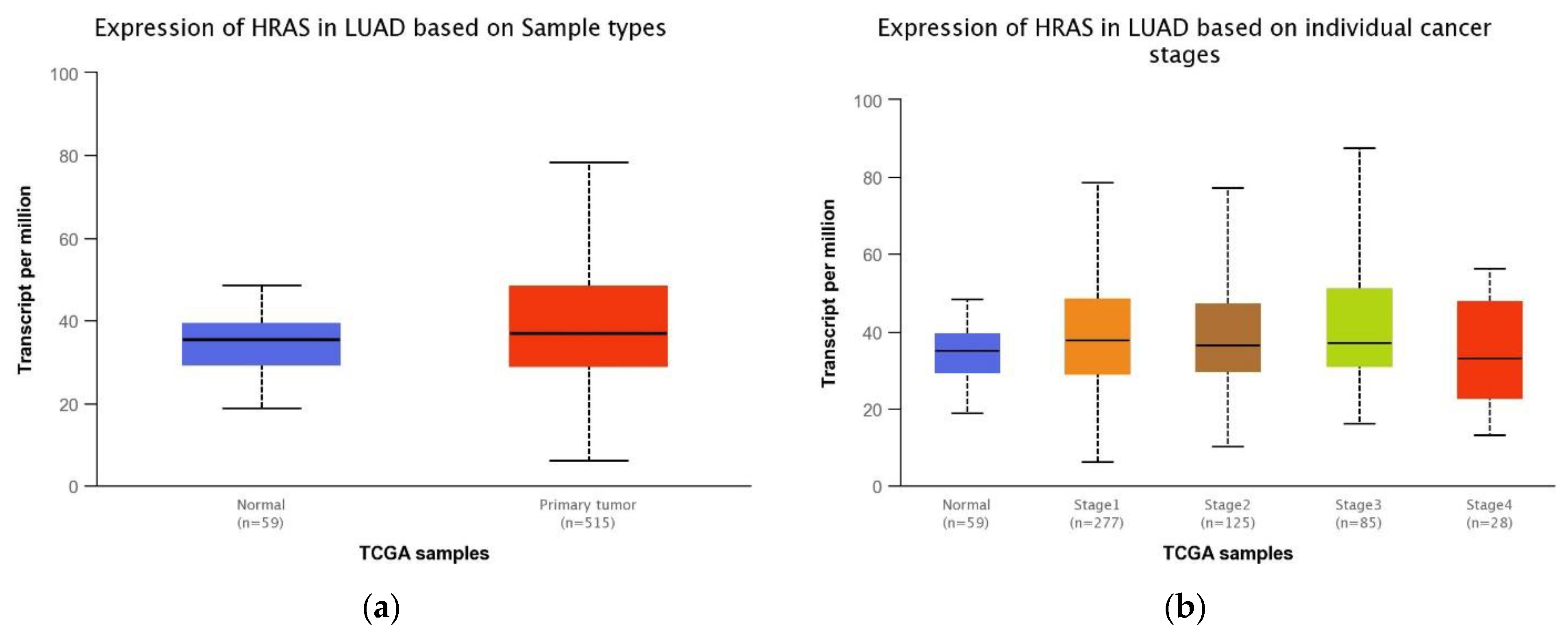
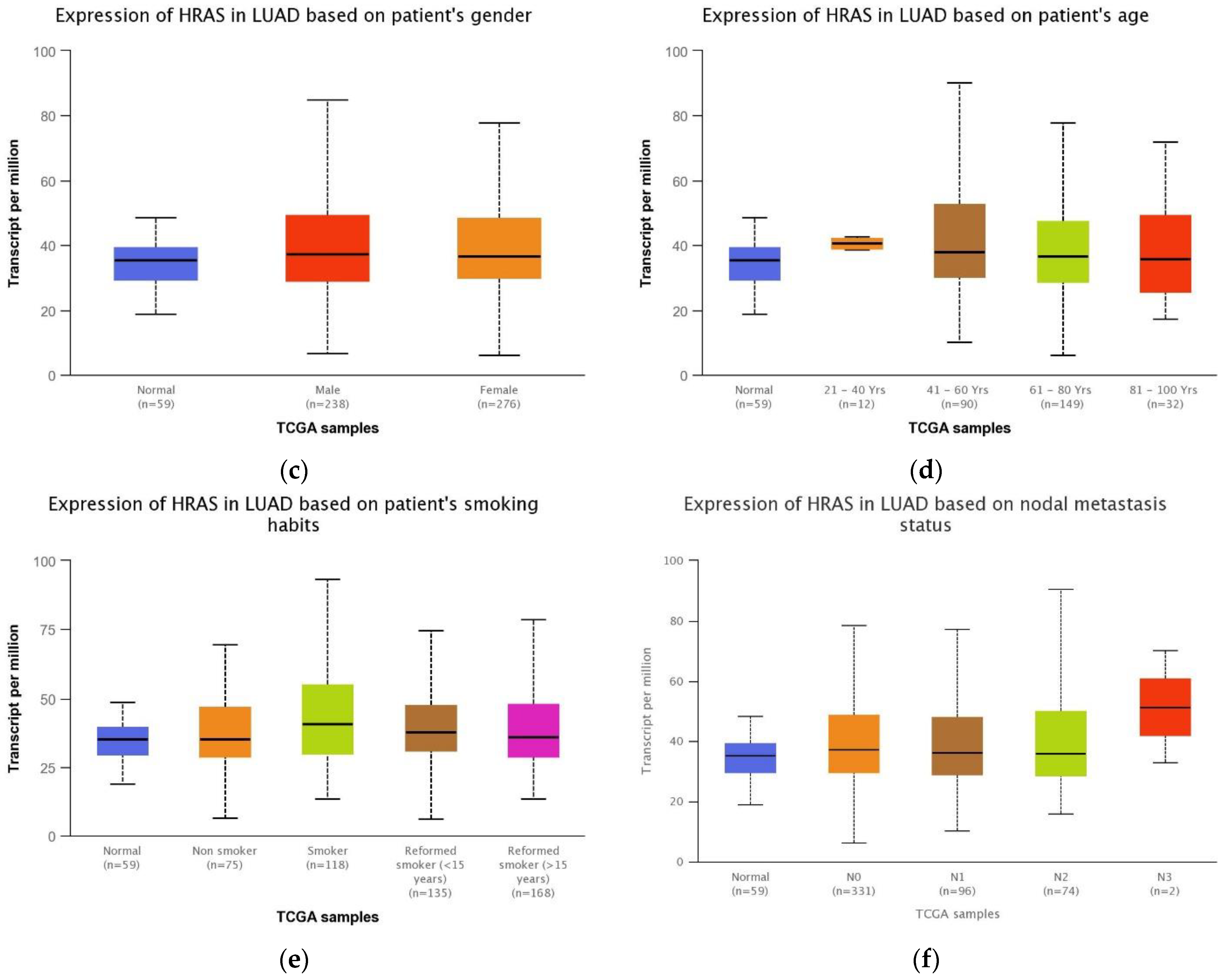
3.5. Association between HRAS Gene Expression and Clinical Features of LUAD Patients
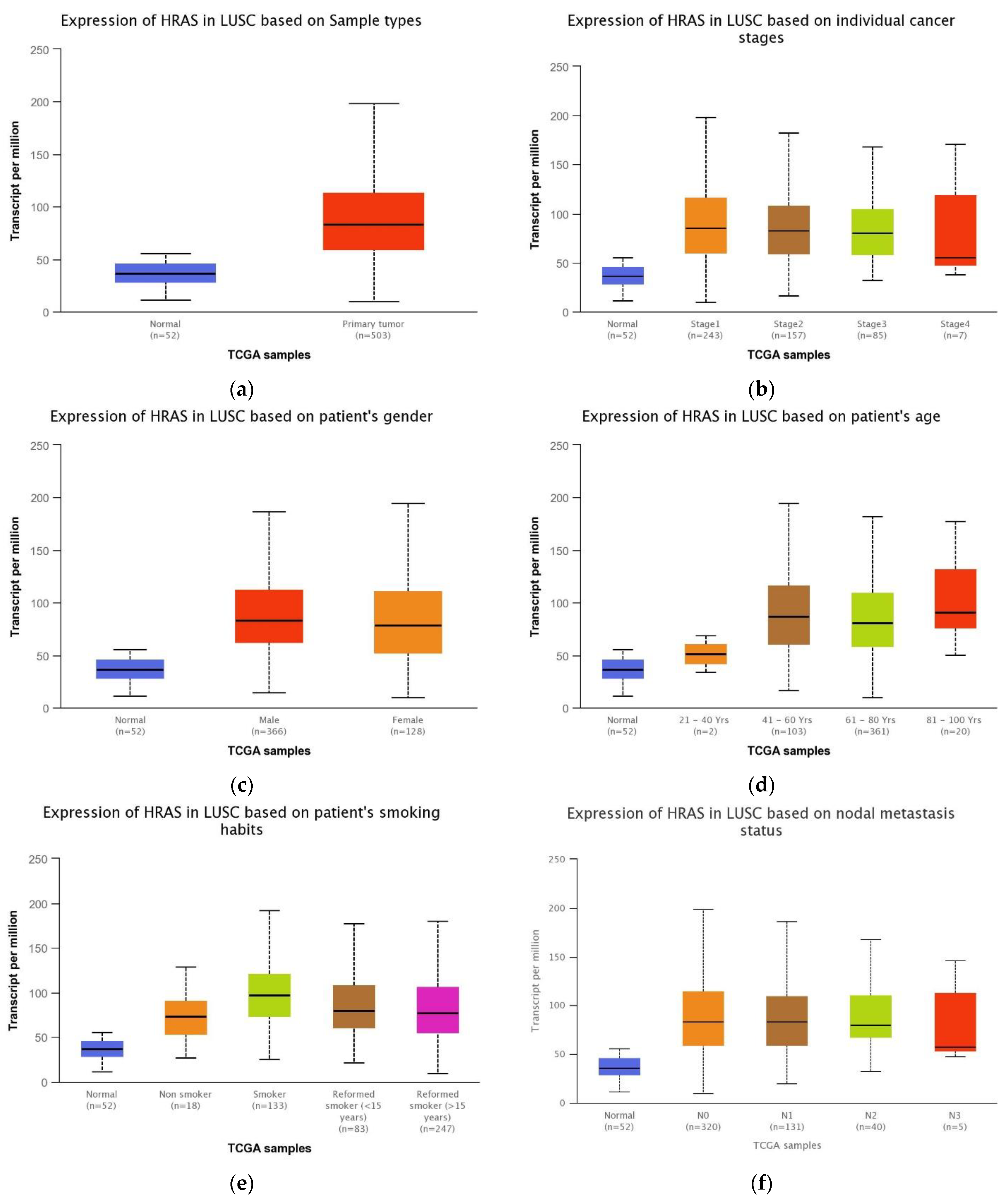
3.6. Association between the HRAS Gene Expression and LUSC Patients’ Clinical Features
3.7. Assessment of the Growth Inhibitory Effects of FTS, Kobe0065 and Deltarasin in A549 NSCLC Cell Line
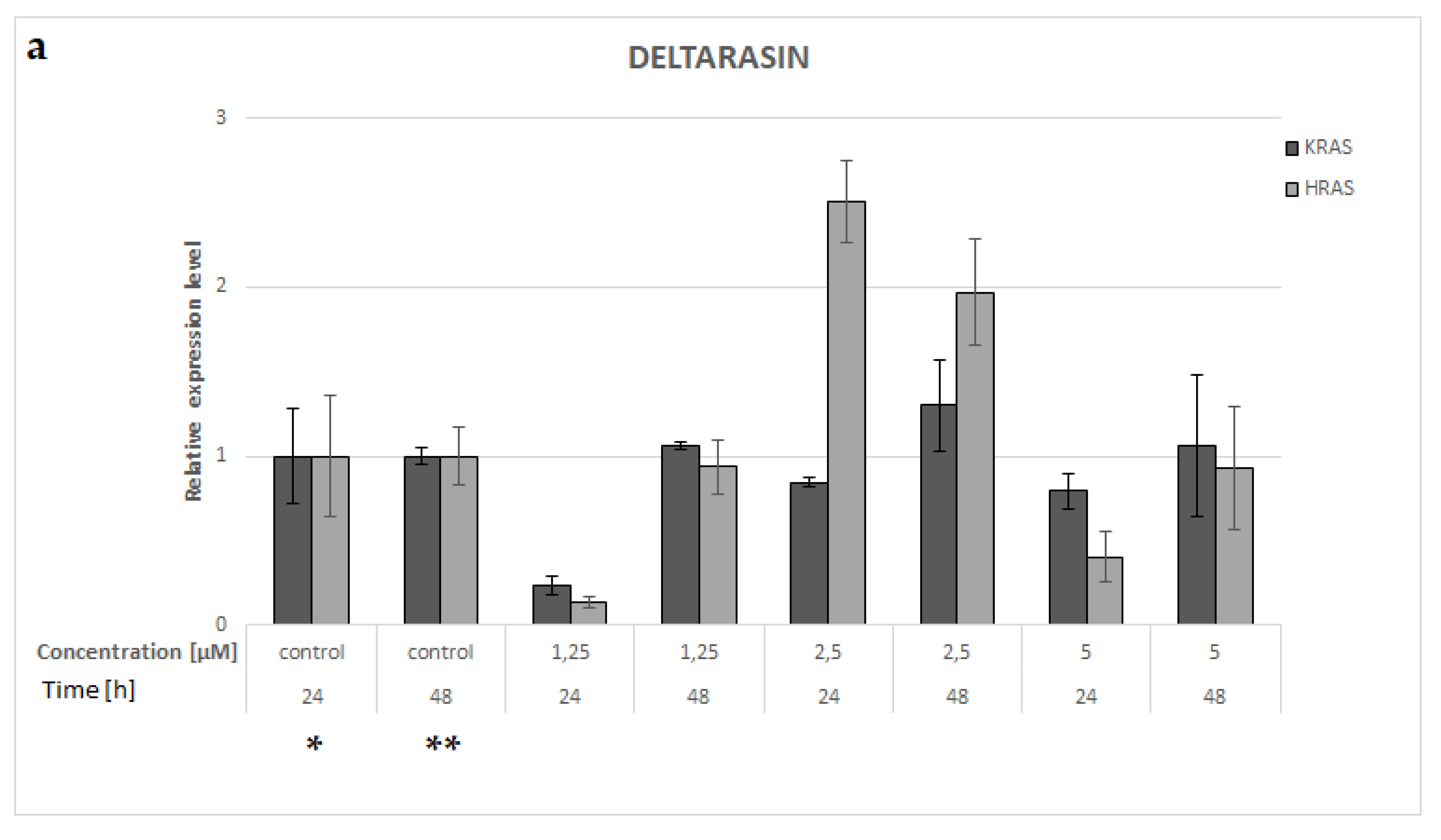
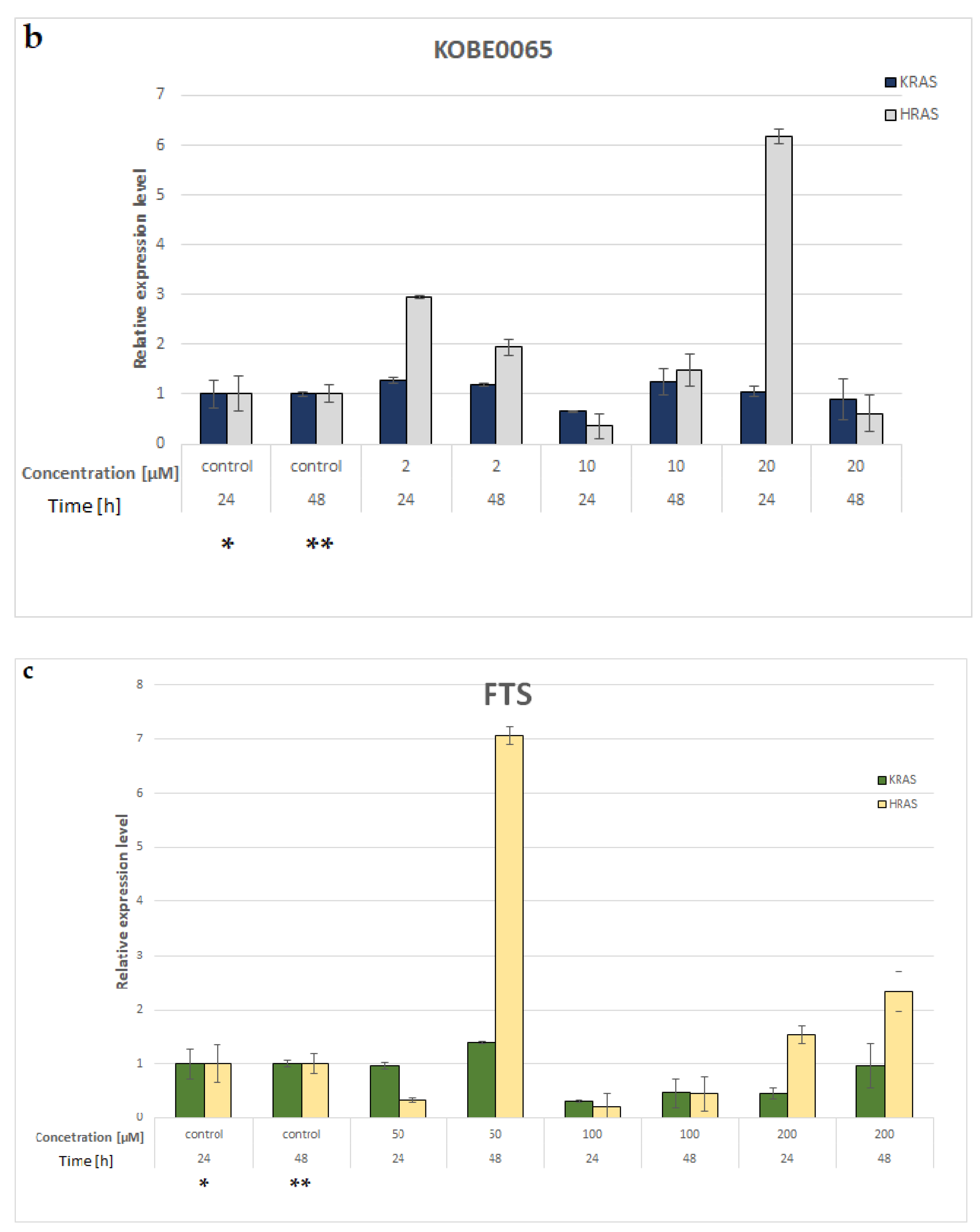
3.8. KRAS and HRAS mRNA Expression in A549 NSCLC Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.B.; Phil, A.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, V.; Omerovic, J. Current Approaches in NSCLC Targeting K-RAS and EGFR. Int. J. Mol. Sci. 2019, 20, 5701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Naemi, H.A.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol. 2019, 54, 1–13. [Google Scholar] [CrossRef]
- Sun, J.-M.; Hwang, D.W.; Ahn, J.S.; Ahn, M.-J.; Park, K. Prognostic and Predictive Value of KRAS Mutations in Advanced Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e64816. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Yang, Y.; Zhu, H.; Zhang, Y.; Zhou, R.; Sun, X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget 2016, 7, 8373–8388. [Google Scholar] [CrossRef] [Green Version]
- Mascaux, C.; De Iannino, N.I.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Haller, A.A.; Lothaire, P.; Meert, A.-P.; Noel, S.E.; et al. The role of RAS oncogene in survival of patients with lung cancer: A systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef]
- Zimmermann, G.; Schultz-Fademrecht, C.; Küchler, P.; Murarka, S.; Ismail, S.; Triola, G.; Nussbaumer, P.; Wittinghofer, A.; Waldmann, H. Structure Guided Design and Kinetic Analysis of Highly Potent Benzimidazole Inhibitors Targeting the PDEδ Prenyl Binding Site. J. Med. Chem. 2014, 57, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Yoshikawa, Y.; Matsumoto, S.; Kataoka, T. Discovery of Small-Molecule Ras Inhibitors that Display Antitumor Activity by Interfering with Ras·GTP–Effector Interaction. Enzymes 2013, 34 Pt B, 1–23. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Lv, H.; Shen, N.; Wang, X.-M.; Tang, S.; Xiong, B.; Ding, J.; Geng, M.-Y.; Huang, M. EPHA2 feedback activation limits the response to PDEδ inhibition in KRAS-dependent cancer cells. Acta Pharmacol. Sin. 2020, 41, 270–277. [Google Scholar] [CrossRef]
- Leung, E.L.H.; Luo, L.X.; Liu, Z.Q.; Wong, V.K.W.; Lu, L.; Xie, Y.; Zhang, N.; Qu, Y.; Fan, X.X.; Li, Y.; et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: Blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Elkon, R.; Yaari, S.; Zundelevich, A.; Jacob-Hirsch, J.; Rechavi, G.; Shamir, R.; Kloog, Y. Gene Expression Signature of Human Cancer Cell Lines Treated with the Ras Inhibitor Salirasib (S-Farnesylthiosalicylic Acid). Cancer Res. 2007, 67, 3320–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Sugita, S.; Enokida, H.; Yoshino, H.; Miyamoto, K.; Yonemori, M.; Sakaguchi, T.; Osako, Y.; Nakagawa, M. HRAS as a potential therapeutic target of salirasib RAS inhibitor in bladder cancer. Int. J. Oncol. 2018, 53, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Arendt, K.A.M.; Ntaliarda, G.; Armenis, V.; Kati, D.; Henning, C.; Giotopoulou, G.A.; Pepe, M.A.A.; Klotz, L.V.; Lamort, A.-S.; Hatz, R.A.; et al. An In Vivo Inflammatory Loop Potentiates KRAS Blockade. Biomedicines 2022, 10, 592. [Google Scholar] [CrossRef]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 73292904, Deltarasin” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Deltarasin (accessed on 14 December 2022).
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 5469318, Salirasib” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Salirasib (accessed on 14 December 2022).
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 3827663” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Kobe0065 (accessed on 14 December 2022).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bontoux, C.; Hofman, V.; Brest, P.; Ilié, M.; Mograbi, B.; Hofman, P. Daily Practice Assessment of KRAS Status in NSCLC Patients: A New Challenge for the Thoracic Pathologist Is Right around the Corner. Cancers 2022, 14, 1628. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Kendzia, B.; Gustavsson, P.; Jöckel, K.-H.; Johnen, G.; Pohlabeln, H.; Olsson, A.; Ahrens, W.; Gross, I.M.; Brüske, I.; et al. Cigarette smoking and lung cancer-relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 2011, 131, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Aran, V.; Domingues, P.M.; de Macedo, F.C.; de Sousa, C.A.M.; Montella, T.C.; Accioly, M.T.D.S.; Gil Ferreira, C. A cross-sectional study examining the expression of splice variants K-RAS4A and K-RAS4B in advanced non-small-cell lung cancer patients. Lung Cancer 2018, 116, 7–14. [Google Scholar] [CrossRef]
- Pązik, M.; Michalska, K.; Żebrowska-Nawrocka, M.; Zawadzka, I.; Łochowski, M.; Balcerczak, E. Clinical significance of HRAS and KRAS genes expression in patients with non–small-cell lung cancer—Preliminary findings. BMC Cancer 2021, 21, 130. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, J.; Shao, C.; Zhao, L.; Xu, W.; Sutherland, L.C.; Wang, K. Differential Expression of RBM5, EGFR and KRAS mRNA and protein in non-small cell lung cancer tissues. J. Exp. Clin. Cancer Res. 2012, 31, 36–39. [Google Scholar] [CrossRef] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lin, Z. Non-Small Cell Lung Cancer Targeted Therapy: Drugs and Mechanisms of Drug Resistance. Int. J. Mol. Sci. 2022, 23, 15056. [Google Scholar] [CrossRef]
- Cascetta, P.; Marinello, A.; Lazzari, C.; Gregorc, V.; Planchard, D.; Bianco, R.; Normanno, N.; Morabito, A. KRAS in NSCLC: State of the Art and Future Perspectives. Cancers 2022, 14, 5430. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Elad-Sfadia, G.; Haklai, R.; Kloog, Y. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Mol. Cancer Ther. 2007, 6, 1765–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, X.; Tang, H.; Cheng, M.; Yang, F.; Xu, W. Design, synthesis, and biological evaluation of novel substituted thiourea derivatives as potential anticancer agents for NSCLC by blocking K-Ras protein-effectors interactions. J. Enzym. Inhib. Med. Chem. 2020, 35, 344–353. [Google Scholar] [CrossRef]
- Eng, S.-K.; Imtiaz, I.; Goh, B.-H.; Ming, L.; Lim, Y.-C.; Lee, W.-L. Does KRAS Play a Role in the Regulation of Colon Cancer Cells-Derived Exosomes? Biology 2021, 10, 58. [Google Scholar] [CrossRef]
- Charette, N.; De Saeger, C.; Lannoy, V.; Horsmans, Y.; Leclercq, I.; Stärkel, P. Salirasib inhibits the growth of hepatocarcinoma cell lines in vitro and tumor growth in vivo through ras and mTOR inhibition. Mol. Cancer 2010, 9, 256. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Brownstein, M.; Haklai, R.; Kloog, Y. Downregulation of survivin and aurora A by histone deacetylase and RAS inhibitors: A new drug combination for cancer therapy. Int. J. Cancer 2011, 128, 691–701. [Google Scholar] [CrossRef]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W.; et al. A Phase II Trial of Salirasib in Patients with Lung Adenocarcinomas with KRAS Mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef] [Green Version]
- Uras, I.Z.; Moll, H.P.; Casanova, E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 4325. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | GeneBank Accesion Number | Sequence | Product Length (bp) |
|---|---|---|---|
| GAPDH | NM_001289745 | Reverse: 5′-ATGCCAGTGAGCTTCCCGTTCAGC-3’ | 145 |
| Forward: 5′-TGGTATCGTGGAAGGACTCATGAC-3’ | |||
| HRAS | NM_176795 | Reverse: 5′-GCCTGGCCCCACCTGTG-3’ | 276 |
| Forward: 5′-CACGGAAGGTCCTGAGGGG-3’ | |||
| KRAS | NM_004985 | Reverse: 5′-TCCTGTAGGAATCCTCTATTG-3’ | 134 |
| Forward: 5′-GCCTGCTGAAAATGACTG-3’ |
| Compound | Concentration [µM] | Cell Viability 24 h [%] | Cell Viability 48 h [%] |
|---|---|---|---|
| Deltarasin | 1.25 | 88.56 | 100 |
| 2.5 | 98.98 | 100 | |
| 5 | 96.87 | 92.7 | |
| Kobe0065 | 2 | 84.67 | 100 |
| 10 | 90.53 | 100 | |
| 20 | 95.1 | 100 | |
| FTS | 50 | 93.29 | 100 |
| 100 | 95.8 | 77 | |
| 200 | 98.12 | 11.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pązik, M.; Żebrowska-Nawrocka, M.; Wosiak, A.; Pietrzak, J.; Balcerczak, E. Comprehensive Analysis of the Expression and Clinical Significance of RAS Family Members in Non-Small Cell Lung Cancer Based on Bioinformatics Data and the A549 Cell Line Model. Appl. Sci. 2023, 13, 166. https://doi.org/10.3390/app13010166
Pązik M, Żebrowska-Nawrocka M, Wosiak A, Pietrzak J, Balcerczak E. Comprehensive Analysis of the Expression and Clinical Significance of RAS Family Members in Non-Small Cell Lung Cancer Based on Bioinformatics Data and the A549 Cell Line Model. Applied Sciences. 2023; 13(1):166. https://doi.org/10.3390/app13010166
Chicago/Turabian StylePązik, Milena, Marta Żebrowska-Nawrocka, Agnieszka Wosiak, Jacek Pietrzak, and Ewa Balcerczak. 2023. "Comprehensive Analysis of the Expression and Clinical Significance of RAS Family Members in Non-Small Cell Lung Cancer Based on Bioinformatics Data and the A549 Cell Line Model" Applied Sciences 13, no. 1: 166. https://doi.org/10.3390/app13010166