
Metabolite Profiling of Christia vespertilionis Leaf Metabolome via Molecular Network Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Collection and Sample Preparation
2.3. Extraction
2.4. UHPLC-MS/MS Analysis
2.5. Generation of Molecular Network
2.6. Semiquantitative Analysis
2.7. Fractionation and MS-Targeted Isolation of Compounds 28 and 47
3. Results and Discussion
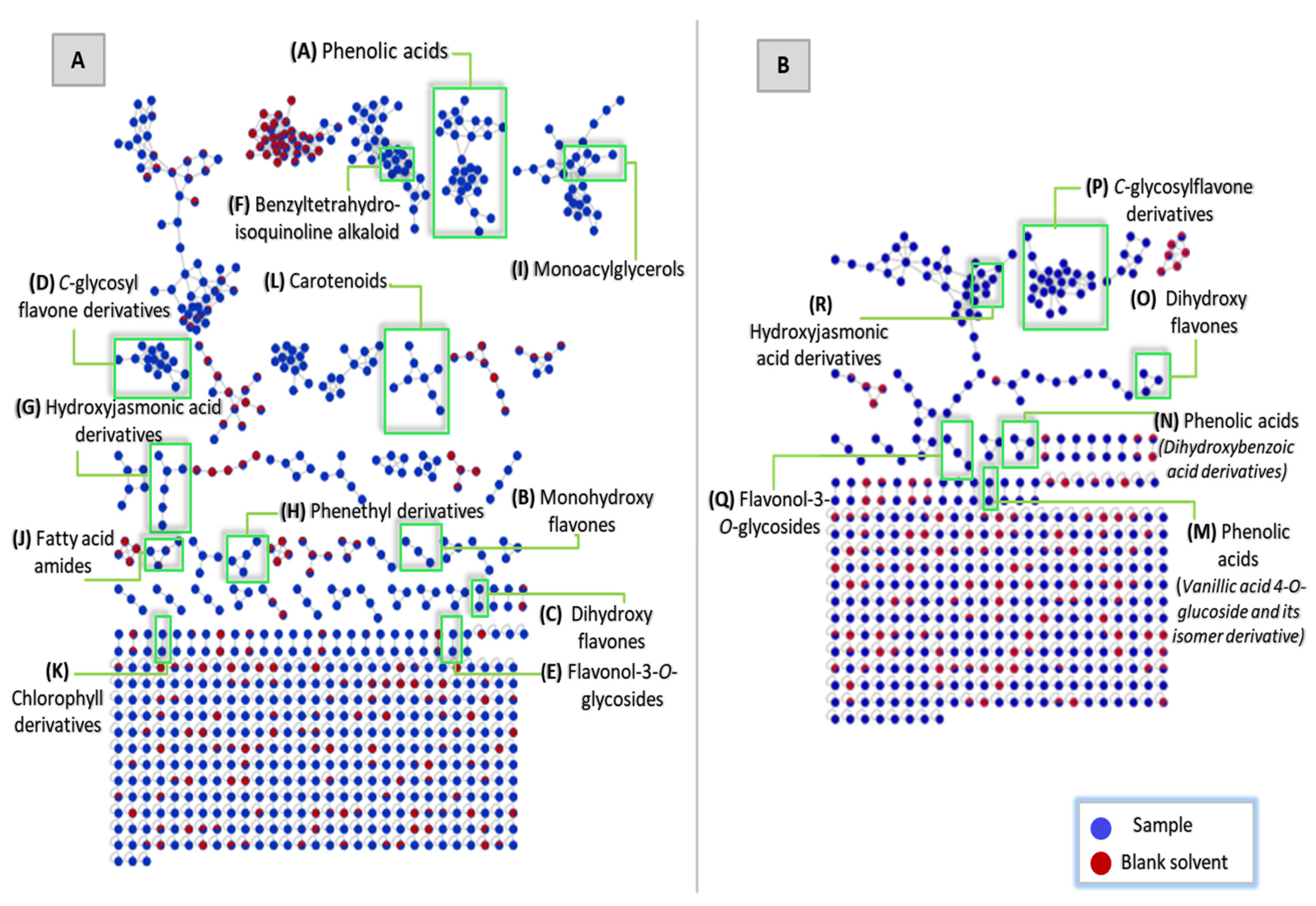
3.1. Metabolite Profiling of the Leaf Metabolome of the Green-Leafed Variety of Christia vespertilionis via Untargeted Tandem Mass Spectrometry-Based Molecular Networking
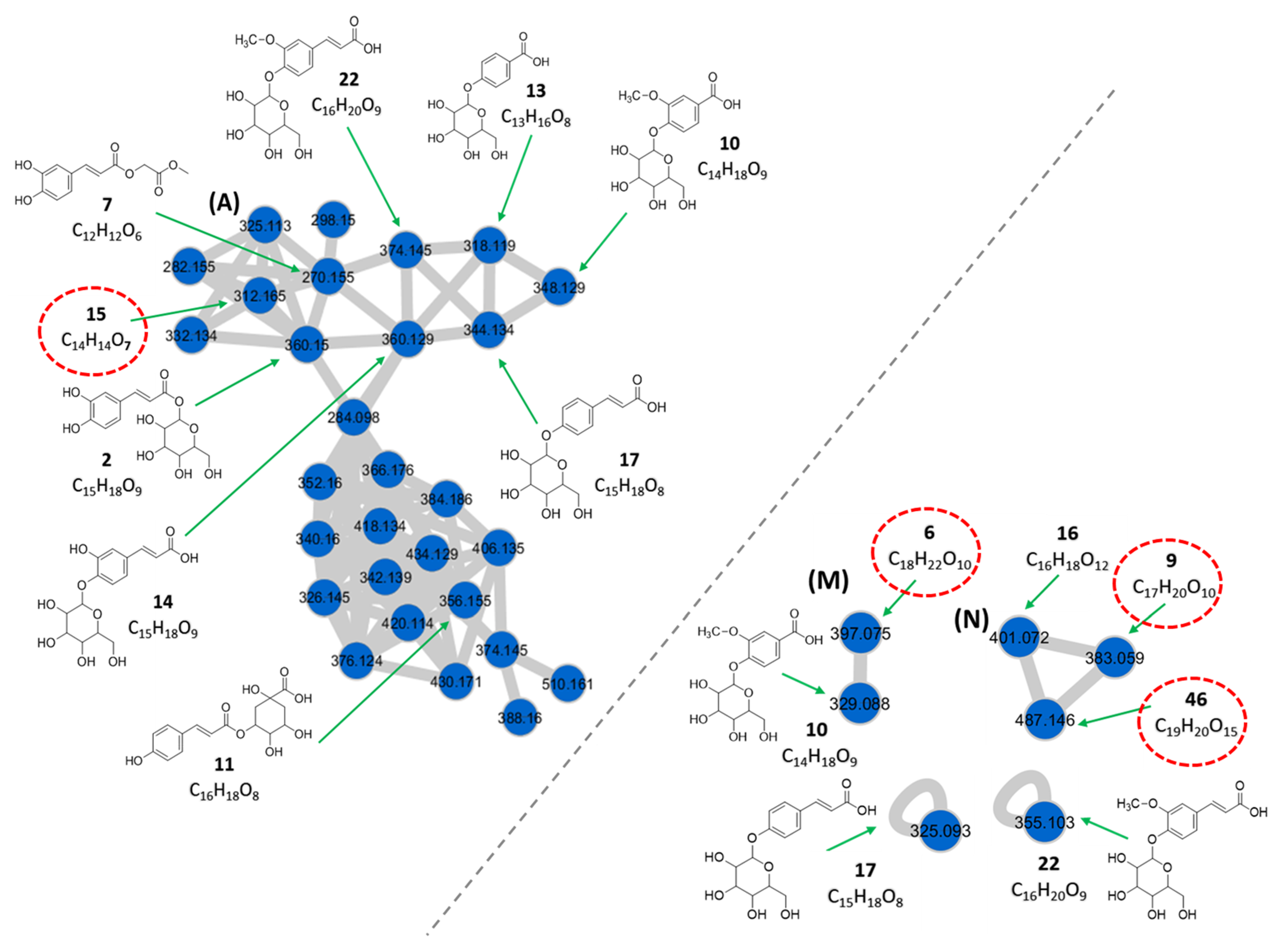
3.1.1. Phenolic Acids
3.1.2. Flavonoids
Monohydroxyflavones
Dihydroxyflavones
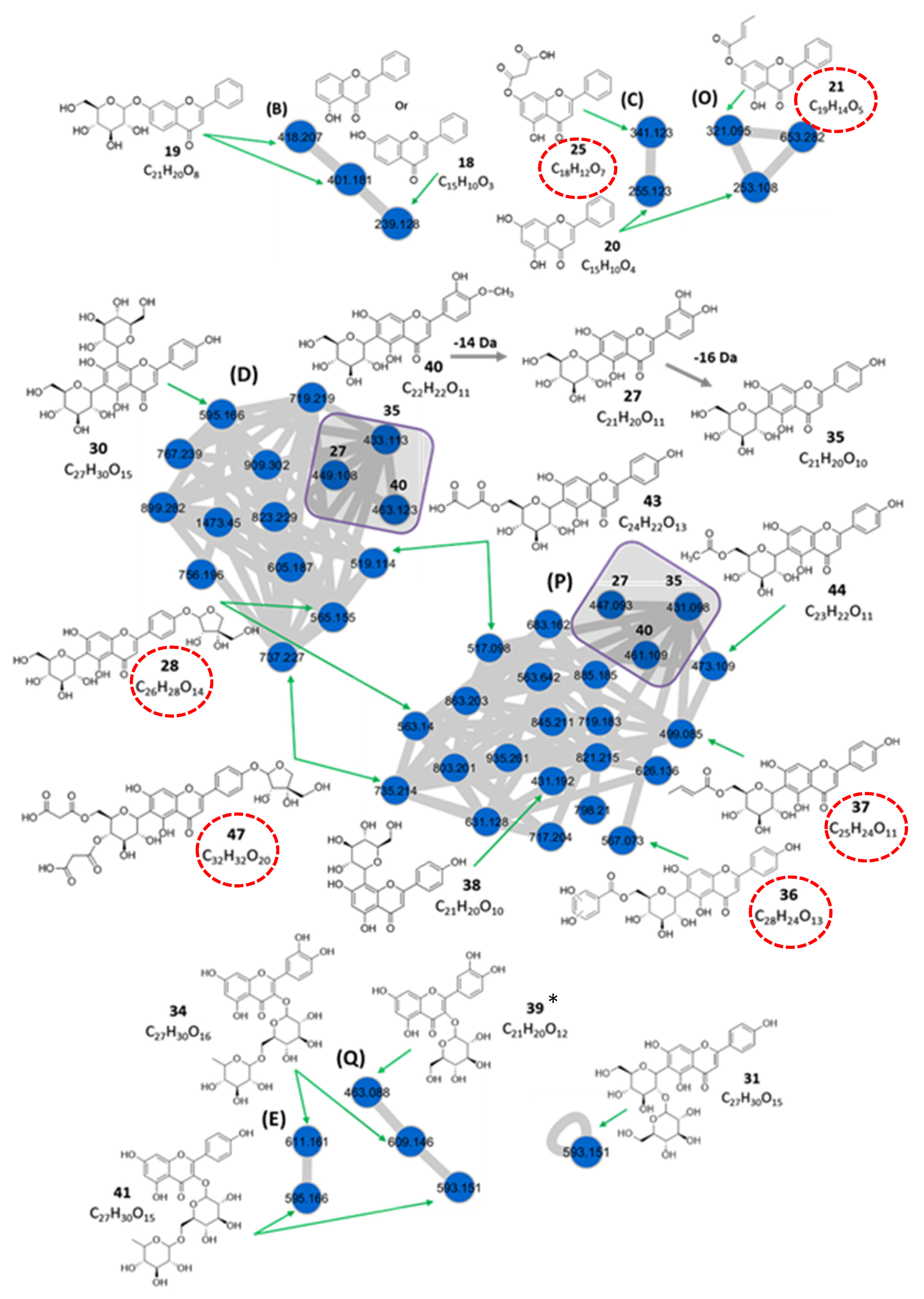
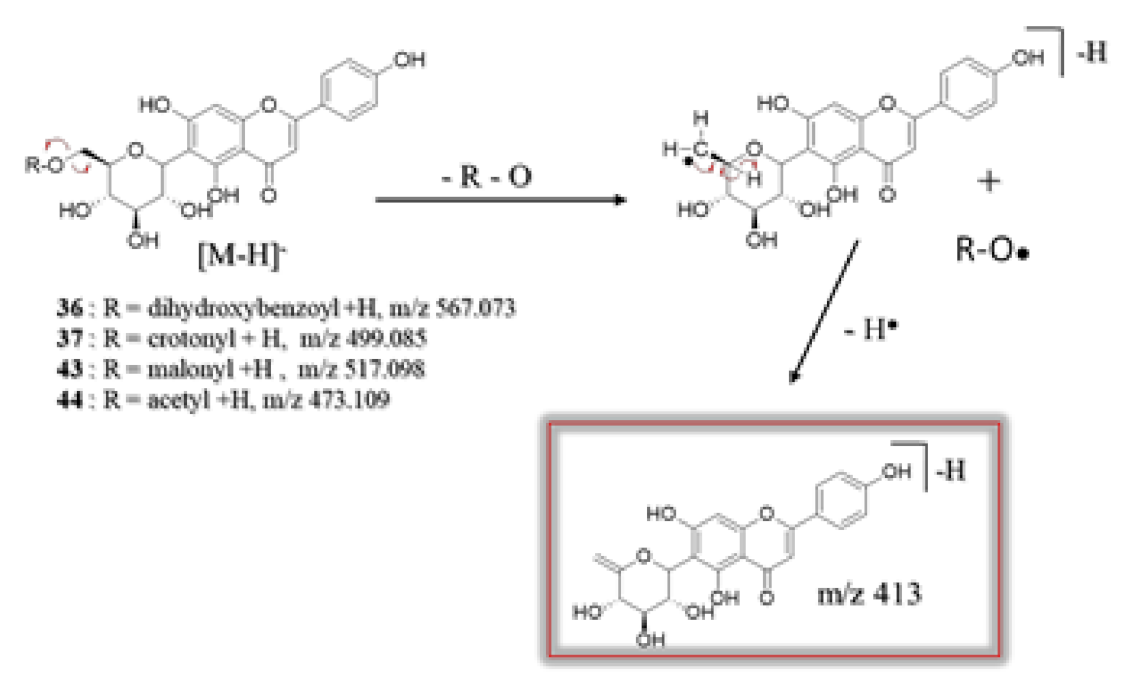
C-glycosylflavone Derivatives
Flavone-C,O-diglycoside
Flavonol-3-O-glycosides
3.1.3. Benzyltetrahydroisoquinoline Alkaloids
3.1.4. Hydroxyjasmonic Acid Derivatives
3.1.5. Phenethyl Derivatives
3.1.6. Monoacylglycerols
3.1.7. Fatty Acid Amides
3.1.8. Chlorophyll Derivatives
3.1.9. Carotenoids
3.1.10. Other Compounds
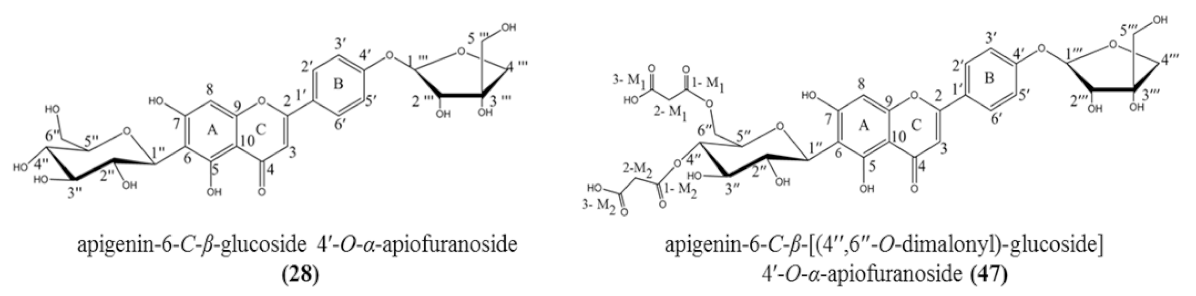
3.2. Structural Elucidation of Isolated Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ismail, S.; Mokhtar, S. The antecedents of herbal product actual purchase in Malaysia. Manag. Sci. Lett. 2015, 5, 771–780. [Google Scholar] [CrossRef]
- Hudson, A.; Lopez, E.; Almalki, A.J.; Roe, A.L.; Calderón, A.I. A Review of the Toxicity of Compounds Found in Herbal Dietary Supplements. Planta Med. 2018, 84, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Aziz, Z.; Tey, N.P. Herbal medicines: Prevalence and predictors of use among Malaysian adults. Complement. Ther. Med. 2009, 17, 44–50. [Google Scholar] [CrossRef]
- Dash, G. An appraisal of Christia vespertilionis (L.F.) bakh. F.: A promising medicinal plant. Int. J. Pharmacogn. Phytochem. Res. 2016, 8, 1037–1039. [Google Scholar]
- Hofer, D.; Schwach, G.; Tabrizi-Wizsy, N.G.; Sadjak, A.; Sturm, S.; Stuppner, H.; Pfragner, R. Christia vespertilionis plant extracts as novel antiproliferative agent against human neuroendocrine tumor cells. Oncol. Rep. 2013, 29, 2219–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen-Pouplin, J.; Tran, H.; Tran, H.; Phan, T.A.; Dolecek, C.; Farrar, J.; Tran, T.H.; Caron, P.; Bodo, B.; Grellier, P. Antimalarial and cytotoxic activities of ethnopharmacologically selected medicinal plants from South Vietnam. J. Ethnopharmacol. 2007, 109, 417–427. [Google Scholar] [CrossRef]
- Abd Mutalib, N.; Abd Latip, N. Synergistic Interactions between Christia Vespertilionis Leaves Extract and Chemotherapy Drug Cyclophosphamide on WRL-68 Cell Line. Asian J. Pharm. Res. Dev. 2019, 7, 109–113. [Google Scholar]
- Lee, J.J.; Saiful Yazan, L.; Kassim, N.K.; Che Abdullah, C.A.; Esa, N.; Lim, P.C.; Tan, D.C. Cytotoxic Activity of Christia vespertilionis Root and Leaf Extracts and Fractions against Breast Cancer Cell Lines. Molecules 2020, 25, 2610. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, H.C.; Sisodia, B.S.; Cheema, H.S.; Agrawal, J.; Pal, A.; Darokar, M.P.; Srivastava, S.K. Novel Antiplasmodial Agents from Christia vespertilionis. Nat. Prod. Commun. 2013, 8, 1591–1594. [Google Scholar] [CrossRef] [Green Version]
- Murugesu, S.; Perumal, V.; Balan, T.; Fatinanthan, S.; Khatib, A.; Arifin, N.J.; Shukri, N.S.S.M.; Saleh, M.S.M.; Hin, L.W. The investigation of antioxidant and antidiabetic activities of Christia vespertilionis leaves extracts. S. Afr. J. Bot. 2020, 133, 227–235. [Google Scholar] [CrossRef]
- Mohd Yasin, Z.N.; Zakaria, M.A.; Nik Mat Zin, N.N.I.; Ibrahim, N.; Mohamad, F.S.; Mat Desa, W.N.S.; Sul’ain, M.D.; Abu Bakar, N. Biological activities and GCMS analysis of the methanolic extract of Christia vespertilionis (L.F.) Bakh. F. leaves. Asian J. Med. Biomed. 2020, 4, 78–88. [Google Scholar] [CrossRef]
- Aguilar-mogas, A.; Sales-pardo, M.; Navarro, M.; Guimerà, R.; Yanes, O. iMet: A network-based computational tool to assist in the annotation of metabolites from tandem mass spectra. Anal. Chem. 2017, 89, 3474–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S.; de Vos, R.C.H.; van Schaik, R.; Vervoort, J. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 2012, 26, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S. Automatic Compound Annotation from Mass Spectrometry Data Using MAGMa. Mass Spectrom. 2014, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Allard, P.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaidon, M.; Wolfender, J. Integration of Molecular Networking and In-Silico MS/MS Fragmentation for Natural Products Dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef]
- Quinn, R.A.; Nothias, L.F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular Networking as a Drug Discovery, Drug Metabolism, and Precision Medicine Strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular Networking as a Dereplication Strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.M.; Lee, S.S.; Chung, B.Y.; Cho, J.-Y.; Lee, I.C.; Ahn, S.R.; Jang, S.J.; Kim, T.H. Pancreatic Lipase Inhibition by C-Glycosidic Flavones Isolated from Eremochloa ophiuroides. Molecules 2010, 15, 8251–8259. [Google Scholar] [CrossRef] [Green Version]
- Hirota, B.C.K.; Miyazaki, C.M.S.; Mercali, C.A.; Verdan, M.C.; Kalegari, M.; Gemin, C.; Lordello, A.L.L.; Miguel, M.D.; Miguel, O.G. C-glycosyl flavones and a comparative study of the antioxidant, hemolytic and toxic potential of Jatropha multifida leaves and bark. Int. J. Phytomed. 2012, 4, 1–5. [Google Scholar]
- Mekky, R.H.; del Contreras, M.M.; El-Gindi, M.R.; Abdel-Monem, A.R.; Abdel-Sattar, E.; Segura-Carretero, A. Profiling of phenolic and other compounds from Egyptian cultivars of chickpea (Cicer arietinum L.) and antioxidant activity: A comparative study. RSC Adv. 2015, 5, 17751–17767. [Google Scholar] [CrossRef]
- Cuyckens, F.; Claeys, M. Mass spectrometry in the structural analysis of flavonoids. J. Mass Spectrom. 2004, 39, 1–15. [Google Scholar] [CrossRef]
- Tsimogiannis, D.; Samiotaki, M.; Panayotou, G.; Oreopoulou, V. Characterization of Flavonoid Subgroups and Hydroxy Substitution by HPLC-MS/MS. Molecules 2007, 12, 593–606. [Google Scholar] [CrossRef]
- El Sayed, A.M.; Ezzat, S.M.; El Naggar, M.M.; El Hawary, S.S. In vivo diabetic wound healing effect and HPLC–DAD–ESI–MS/MS profiling of the methanol extracts of eight Aloe species. Rev. Bras. Farmacogn. 2016, 26, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Abad-García, B.; Garmón-Lobato, S.; Berrueta, L.A.; Gallo, B.; Vicente, F. New features on the fragmentation and differentiation of C-glycosidic flavone isomers by positive electrospray ionization and triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 1834–1842. [Google Scholar] [CrossRef]
- Negri, G.; Santi, D.D.; Tabach, R. Chemical composition of hydroethanolic extracts from Siparuna guianensis, medicinal plant used as anxiolytics in Amazon region. Rev. Bras. Farmacogn. 2012, 22, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Kachlicki, P.; Piasecka, A.; Stobiecki, M.; Marczak, L. Structural Characterization of Flavonoid Glycoconjugates and Their Derivatives with Mass Spectrometric Techniques. Molecules 2016, 21, 1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yu, H.; Wu, H.; Pan, Y.; Wang, K.; Jin, Y.; Zhang, C. Characterization and Quantification by LC-MS/MS of the Chemical Components of the Heating Products of the Flavonoids Extract in Pollen Typhae for Transformation Rule Exploration. Molecules 2015, 20, 18352–18366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiting, P.A. Commercial Production of Christia subcordata Moench by Establishing Cultural Practices and by Applying Plant Growth Regulators. The University of Georgia, 2007. Available online: http://athenaeum.libs.uga.edu (accessed on 25 September 2020).
- Wink, M. Evolution of secondary metabolites in legumes (Fabaceae). S. Afr. J. Bot. 2013, 89, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Qing, Z.; Xu, Y.; Yu, L.; Liu, J.; Huang, X.; Tang, Z.; Cheng, P.; Zeng, J. Investigation of fragmentation behaviours of isoquinoline alkaloids by mass spectrometry combined with computational chemistry. Sci. Rep. 2020, 10, 733. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Chandra, P.; Bajpai, V.; Singh, A.; Srivastava, M.; Mishra, D.K.; Kumar, B. Rapid qualitative and quantitative analysis of bioactive compounds from Phyllanthus amarus using LC/MS/MS techniques. Ind. Crop. Prod. 2015, 69, 143–152. [Google Scholar] [CrossRef]
- Murakami, T.; Kohno, K.; Ninomiya, K.; Matsuda, H.; Yoshikawa, M. Medicinal Foodstuffs. XXV. Hepatoprotective Principle and Structures of Ionone Glucoside, Phenethyl Glycoside, and Flavonol Oligoglycosides from Young Seedpods of Garden Peas, Pisum sativum L. Chem. Pharm. Bull. 2001, 49, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Chua, Y.G.; Bloodworth, B.C.; Leong, L.P.; Li, S.F.Y. Metabolite profiling of edible bird’s nest using gas chromatography/mass spectrometry and liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Shioi, Y.; Watanabe, K.; Takamiya, K.-I. Enzymatic Conversion of Pheophorbide a to the Precursor of Pyropheophorbide a in Leaves of Chenopodium album. Plant Cell Physiol. 1996, 37, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.M.; Christou, P.; Canela-Garayoa, R. Identification of carotenoids using mass spectrometry. Mass Spectrom. Rev. 2013, 33, 353–372. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, M.; Tang, D.; Shi, Y. Composition of Carotenoids and Flavonoids in Narcissus Cultivars and their Relationship with Flower Color. PLoS ONE 2015, 10, e0142074. [Google Scholar] [CrossRef] [Green Version]
- van Breemen, R.B.; Dong, L.; Pajkovic, N.D. Atmospheric pressure chemical ionization tandem mass spectrometry of carotenoids. Int. J. Mass Spectrom. 2012, 312, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Lin, Z.; Jiang, H.; Tong, L.; Wang, H.; Chen, S. Rapid Identification and Assignation of the Active Ingredients in Fufang Banbianlian Injection Using HPLC-DAD-ESI-IT-TOF-MS. J. Chromatogr. Sci. 2016, 54, 1225–1237. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Li, T.; He, M.; Li, Z.; Tan, T.; Zhang, W.; Li, Y.; Feng, Y.; Yang, S. Identification and Quantification Analysis on the Chemical Constituents from Traditional Mongolian Medicine Flos scabiosae Using UHPLC–DAD–Q-TOF-MS Combined with UHPLC–QqQ-MS. J. Chromatogr. Sci. 2016, 54, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Piraud, M.; Vianey-Saban, C.; Petritis, K.; Elfakir, C.; Steghens, J.-P.; Morla, A.; Bouchu, D. ESI-MS/MS analysis of underivatised amino acids: A new tool for the diagnosis of inherited disorders of amino acid metabolism. Fragmentation study of 79 molecules of biological interest in positive and negative ionisation mode. Rapid Commun. Mass Spectrom. 2003, 17, 1297–1311. [Google Scholar] [CrossRef]
- Nikolić, D.; Gödecke, T.; Chen, S.-N.; White, J.; Lankin, D.C.; Pauli, G.F.; van Breemen, R.B. Mass spectrometric dereplication of nitrogen-containing constituents of black cohosh (Cimicifuga racemosa L.). Fitoterapia 2012, 83, 441–460. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Fan, G.; Hong, Z.; Chai, Y.; Wu, Y. Preparative separation of isovitexin and isoorientin from Patrinia villosa Juss by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1074, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Li, J.; Wang, N.-L.; Yao, X.-S. Flavonoids and a New Polyacetylene from Bidens parviflora Willd. Molecules 2008, 13, 1931–1941. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak No. | (min) | Compound Identification | Molecular Formula | Precursor Ion (m/z) | Ion Type | Key Fragments (m/z) | Area | Relative Abundance (%) |
|---|---|---|---|---|---|---|---|---|
| Phenolic acids (Clusters A, M, N, and “self-loop” nodes) | ||||||||
| 2 | 1.03 | Caffeoyl glucoside c | C15H18O9 | 360.150 | [M + NH4]+ | 163, 109 | 2.6 × 108 | 3.99 |
| 6 | 1.54 | Crotonylated derivative of vanillic acid glucosyl ester d** | C18H22O10 | 397.075 | [M − H]− | 329, 122 | 5.3 × 106 | 0.08 |
| 7 | 1.63 | Caffeoylglycolic acid methyl ester c | C12H12O6 | 270.155 | [M + NH4]+ | 163, 109 | 9.4 × 106 | 0.14 |
| 9 | 2.28 | Dihydroxybenzoic acid crotonyl hexoside d** | C17H20O10 | 383.059 | [M − H]− | 315, 153, 152, 109 | 1.3 × 107 | 0.20 |
| 10 | 2.32 | Vanillic acid 4-O-glucoside c | C14H18O9 | 348.129 | [M + NH4]+ | 169, 151 167, 122 | 6.1 × 106 | 0.09 |
| 329.088 | [M − H]− | |||||||
| 11 | 3.05 | p-coumaroylquinic acid c | C16H18O8 | 356.155 | [M + NH4]+ | 175, 131 | 9.4 × 106 | 0.14 |
| 13 | 3.68 | p-hydroxybenzoic acid 4-O-glucoside c | C13H16O8 | 318.119 | [M + NH4]+ | 139, 121 | 5.8 × 106 | 0.09 |
| 14 | 3.89 | Caffeic acid 4-O-glucoside c | C15H18O9 | 360.129 | [M + NH4]+ | 181, 163 | 4.6 × 106 | 0.07 |
| 15 | 3.92 | Isopropyl derivative of caffeoylglycolic acid methyl ester d** | C14H14O7 | 312.165 | [M + NH4]+ | 163, 109 | 3.9 × 106 | 0.06 |
| 16 | 3.96 | Dihydroxybenzoic acid malonyl hexoside b | C16H18O12 | 401.072 | [M − H]− | 357, 315, 153, 152, 109 | 6.7 × 106 | 0.10 |
| 17 | 4.01 | p-coumaric acid 4-O-glucoside a | C15H18O8 | 344.134 | [M + NH4]+ | 165, 147, 119 | 7.2 × 107 | 1.11 |
| 325.093 | [M − H]− | 163 | ||||||
| 22 | 4.48 | Ferulic acid 4-O-glucoside c | C16H20O9 | 374.145 | [M + NH4]+ | 195, 177, 149 193 | 1.0 × 108 | 1.54 |
| 355.103 | [M − H]− | |||||||
| 46 | 8.07 | Dihydroxybenzoic acid dimalonyl hexoside d** | C19H20O15 | 487.146 | [M − H]− | 315, 153, 152, 109 | 6.7 × 106 | 0.10 |
| Monohydroxyflavones (Cluster B) | ||||||||
| 18 | 4.08 | 5 or 7-hydroxyflavone d | C15H10O3 | 239.128 | [M + H]+ | 221, 137, 109, 93 | 2.1 × 107 | 0.32 |
| 19 | 4.11 | 7-hydroxyflavone glucoside c | C21H20O8 | 401.181 | [M + H]+ | 239, 221, 137, 109, 93 239, 221, 137, 109, 93 | 2.0 × 107 | 0.31 |
| 418.207 | [M + NH4]+ | |||||||
| Dihydroxyflavones (Clusters C and O) | ||||||||
| 20 | 4.13 | 5,7-dihydroxyflavone (Chrysin) b | C15H10O4 | 255.123 | [M + H]+ | 237, 209, 177, 153 | 4.9 × 107 | 0.75 |
| 253.108 | [M − H]− | 175, 151 | ||||||
| 21 | 4.13 | 7-O-crotonylchrysin c** | C19H14O5 | 321.095 | [M − H]− | 253, 175, 151 | 5.8 × 106 | 0.09 |
| 25 | 5.09 | 7-O-malonylchrysin c** | C18H12O7 | 341.123 | [M + H]+ | 237, 177, 153 | 4.1 × 106 | 0.06 |
| C-glycosylflavone Derivatives (Clusters D and P) | ||||||||
| 27 | 5.23 | Luteolin-6-C-glucoside (isoorientin) a | C21H20O11 | 449.108 | [M + H]+ | 329, 287 | 5.5 × 107 | 0.84 |
| 447.093 | [M − H]− | 429, 357, 327, 298, 285 | ||||||
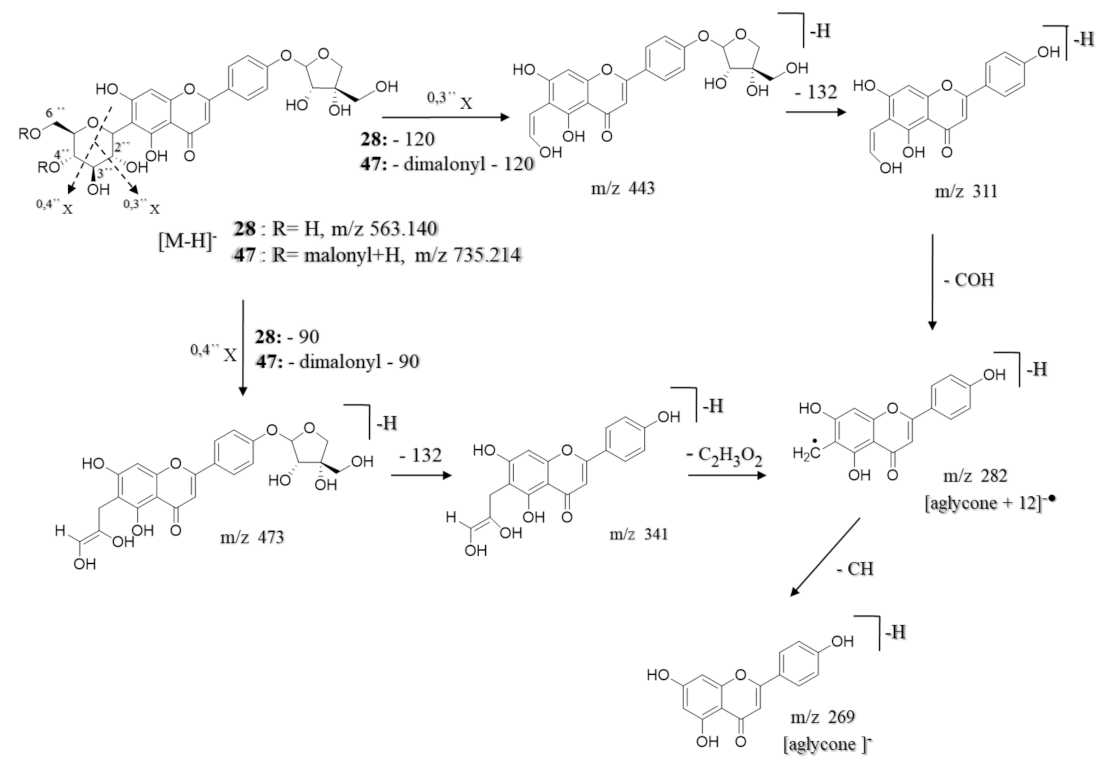
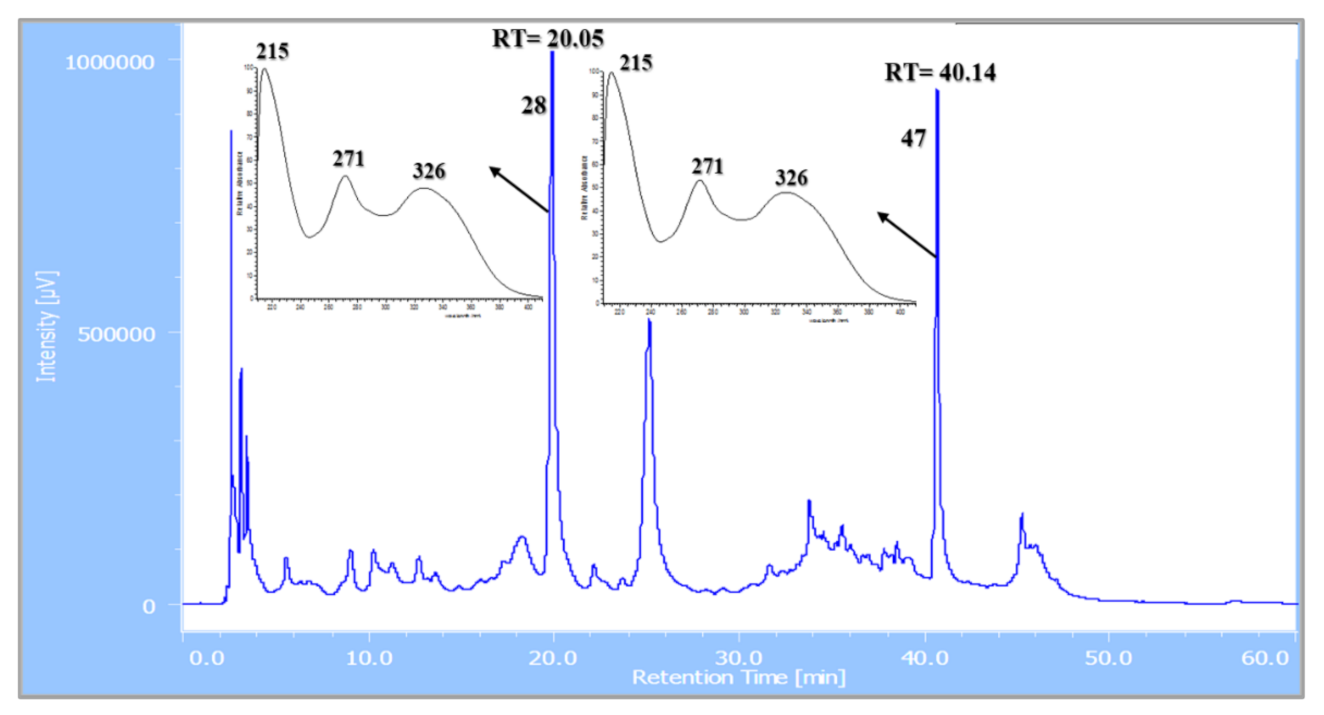
| 28 | 5.35 | Apigenin-6-C-β-glucoside 4′-O-α-apiofuranoside ** | C26H28O14 | 565.155 | [M + H]+ | 445, 313, 284, 271 | 8.7 × 108 | 13.36 |
| 563.140 | [M − H]− | 473, 443, 341, 311, 282, 269 | ||||||
| 30 | 5.60 | Apigenin-6,8-di-C-glucoside (vicenin 2) b | C27H30O15 | 595.166 | [M + H]+ | 433, 337, 367 | 4.2 × 108 | 6.45 |
| 35 | 6.00 | Apigenin-6-C-glucoside (isovitexin) a | C21H20O10 | 433.113 | [M + H]+ | 361, 313, 284, 271 | 7.9 × 108 | 12.13 |
| 431.098 | [M − H]− | 341, 311, 282, 269 | ||||||
| 36 | 6.02 | Apigenin-6-C-(6″-O-dihydroxybenzoyl)- glucoside d** | C28H24O13 | 567.073 | [M − H]− | 413, 341, 311, 282, 269 | 7.2 × 108 | 11.06 |
| 37 | 6.05 | Apigenin-6-C-(6″-O-crotonyl)-glucoside c** | C25H24O11 | 499.085 | [M − H]− | 413, 341, 311, 282, 269 | 6.3 × 108 | 9.67 |
| 38 | 6.17 | Apigenin-8-C-glucoside (vitexin) c | C21H20O10 | 431.192 | [M − H]− | 341, 311, 282, 269 | 3.5 × 107 | 0.54 |
| Peak No. | RT (min) | Compound Identification | Molecular Formula | Precursor Ion (m/z) | Ion Type | Key Fragments (m/z) | Area | Relative Abundance (%) |
| 40 | 6.36 | Diosmetin-6-C-glucoside b | C22H22O11 | 463.123 | [M + H]+ | 391, 343, 313, 297, 151 | 1.9 × 107 | 0.29 |
| 461.109 | [M − H]− | 371, 341, 312, 299 | ||||||
| 43 | 7.13 | Apigenin-6-C-(6″-O-malonyl)-glucoside c | C24H22O13 | 519.114 | [M + H]+ | 415, 313, 271 | 7.4 × 107 | 1.14 |
| 517.098 | [M − H]− | 413, 341, 311, 282, 269 | ||||||
| 44 | 7.13 | Apigenin-6-C-(6″-O-acetyl)-glucoside c | C23H22O11 | 473.109 | [M − H]− | 413, 341, 311, 282, 269 | 5.9 × 106 | 0.09 |
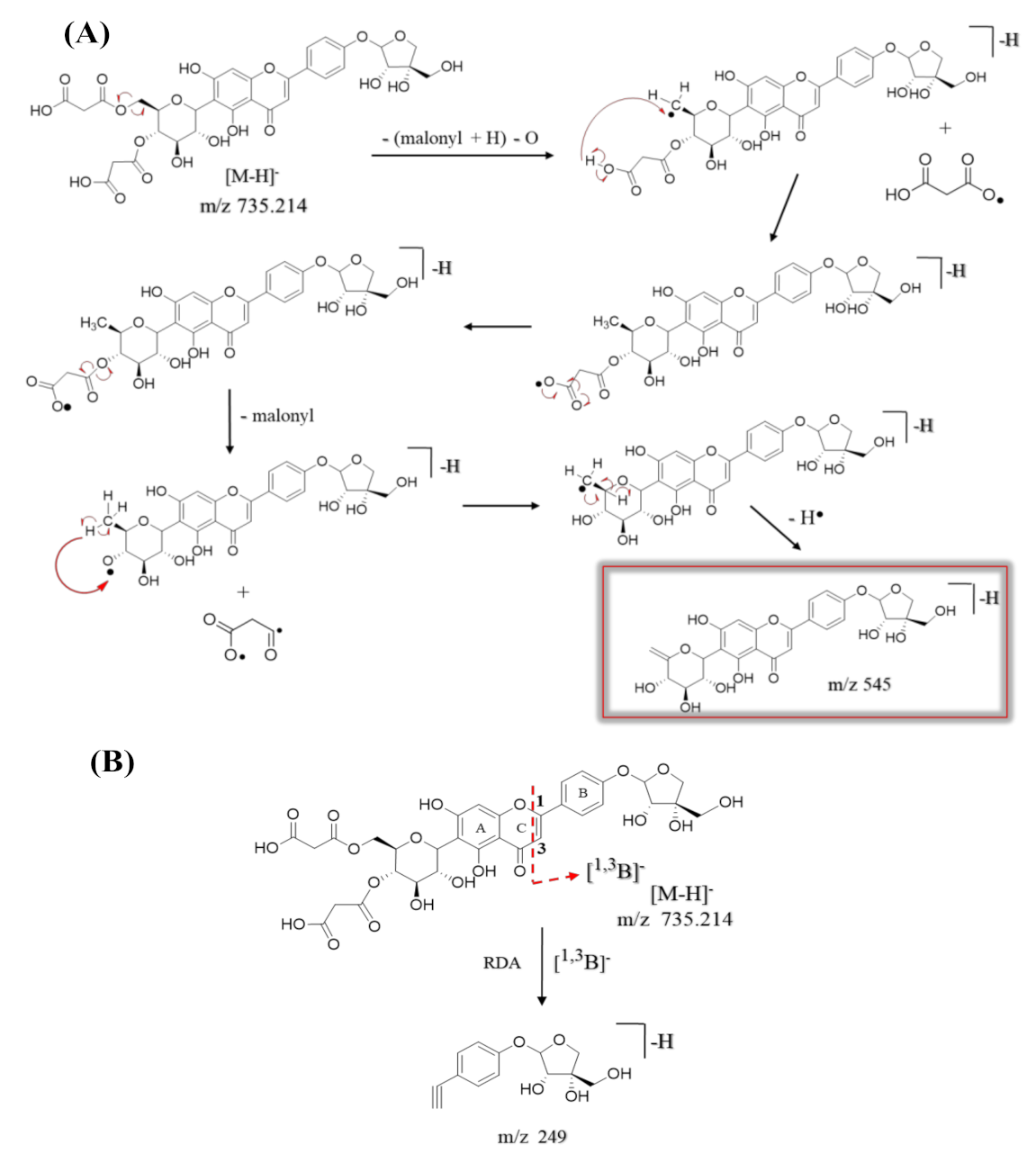
| 47 | 9.36 | Apigenin-6-C-β-[(4″,6″-O-dimalonyl)-glucoside] 4′-O-α-apiofuranoside ** | C32H32O20 | 737.229 | [M + H]+ | 313, 284, 271 | 9.8 × 108 | 15.05 |
| 735.214 | [M − H]− | 545, 473, 443, 341, 311, 282, 269, 249 | ||||||
| Flavone-C,O-diglycoside (“self-loop” node) | ||||||||
| 31 | 5.60 | Apigenin-6-C-glucosyl-2″-O-glucoside (isovitexin-2″-O-glucoside) b | C27H30O15 | 593.151 | [M − H]− | 473, 413, 293 | 1.2 × 108 | 1.84 |
| Flavonol-3-O-glycosides (Clusters E and Q) | ||||||||
| 34 | 5.86 | Quercetin-3-O-rutinoside (Rutin) a | C27H30O16 | 611.161 | [M + H]+ | 465, 303 | 2.1 × 108 | 3.22 |
| 609.146 | [M − H]− | 301, 300, 271, 255, 179, 151 | ||||||
| 39 | 6.21 | Quercetin-3-O-glucoside (Isoquercitrin) a* | C21H20O12 | 463.088 | [M − H]− | 301, 300, 271, 255, 179, 151 | 3.7 × 106 | 0.06 |
| 41 | 6.36 | Kaempferol- 3-O-rutinoside b | C27H30O15 | 595.166 | [M + H]+ | 449, 287 | 4.6 × 107 | 0.71 |
| 593.151 | [M − H]− | 285, 255, 227, 151 | ||||||
| Benzyltetrahydroisoquinoline alkaloids (Cluster F) | ||||||||
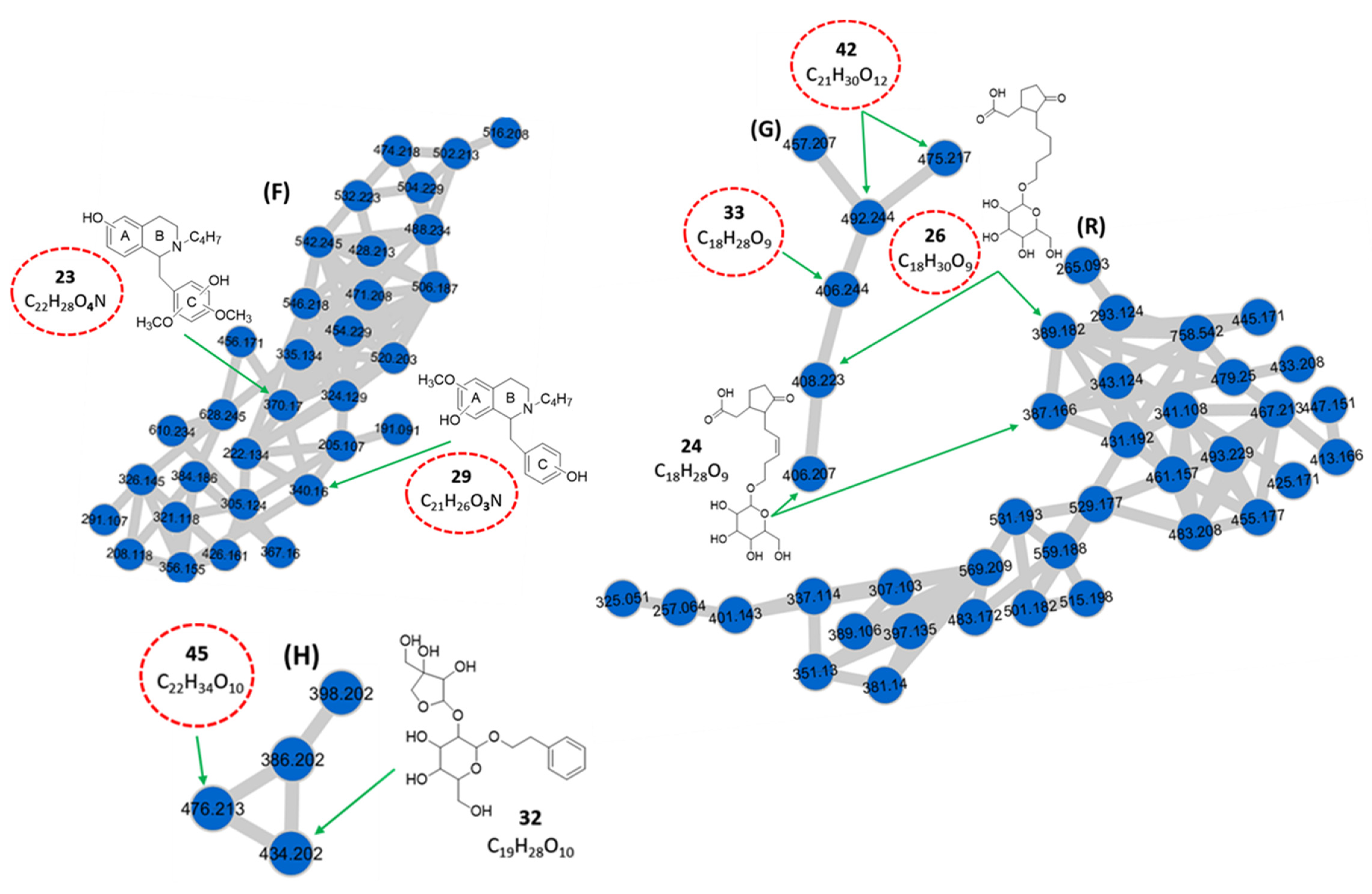
| 23 | 4.51 | Benzyltetrahydroisoquinoline derivative d** | C22H28O4N | 370.170 | [M + H]+ | 299, 145, 127 | 4.9 × 107 | 0.75 |
| 29 | 5.56 | Benzyltetrahydroisoquinoline derivative d** | C21H26O3N | 340.160 | [M + H]+ | 269, 175, 145, 127, 119 | 3.3 × 107 | 0.51 |
| Hydroxyjasmonic acid derivatives (Clusters G and R) | ||||||||
| 24 | 4.71 | 3-oxo-2-[5-(hexopyranosyloxy)-pent-2-enyl]-cyclopentaneacetic acid (Tuberonic acid hexoside) a | C18H28O9 | 406.207 | [M + NH4]+ | 227, 209, 163 | 1.7 × 107 | 0.26 |
| 387.166 | [M − H]− | 207, 59 | ||||||
| 26 | 5.08 | Hydrogenated derivative of tuberonic acid hexoside c** | C18H30O9 | 408.223 | [M + NH4]+ | 229, 211, 165 | 5.7 × 106 | 0.09 |
| 389.182 | [M − H]− | 209, 59 | ||||||
| 33 | 5.75 | Tuberonic acid hexoside isomer d** | C18H28O9 | 406.244 | [M + NH4]+ | 209, 163 | 9.4 × 106 | 0.14 |
| 42 | 6.81 | Malonylated derivative of tuberonic acid hexoside isomer d** | C21H30O12 | 492.244 | [M + NH4]+ | 227, 209, 163 | 7.4 × 106 | 0.11 |
| 475.217 | [M + H]+ | 227, 209, 163 | ||||||
| Phenethyl derivatives (Cluster H) | ||||||||
| 32 | 5.68 | Phenethyl 1-O-β-d-apiofuranosyl (1→2)-β-d-glucopyranoside (Sayaendoside) a | C19H28O10 | 434.202 | [M + NH4]+ | 295, 133, 123 | 5.6 × 106 | 0.09 |
| 45 | 7.56 | Isopropyl derivative of phenethyl 1-O-β-d-apiofuranosyl (1→2)-β-d-glucopyranoside d** | C22H34O10 | 476.213 | [M + NH4]+ | 295, 165, 133 | 8.2 × 106 | 0.13 |
| Monoacylglycerols (Cluster I) | ||||||||
| 48 | 16.66 | 6,9,12,15-Octadecatetraenoic acid, 2,3-dihydroxypropyl ester c | C21H33O3 | 351.254 | [M + H]+ | 259, 241 | 7.5×106 | 0.12 |
| 38 | 6.17 | Apigenin-8-C-glucoside (vitexin) c | C21H20O10 | 431.192 | [M − H]− | 341, 311, 282, 269 | 3.5 × 107 | 0.54 |
| Peak No. | RT (min) | Compound Identification | Molecular Formula | Precursor Ion (m/z) | Ion Type | Key Fragments (m/z) | Area | Relative Abundance (%) |
| 49 | 19.17 | 9,12,15-Octadecatrienoic acid, 3-(hexopyranosyl)-2-hydroxylpropyl ester a | C27H46O9 | 532.348 | [M + NH4]+ | 353, 261, 243 | 6.8 × 106 | 0.10 |
| 50 | 22.11 | 9,12,15-Octadecatrienoic acid, 2,3-dihydroxypropyl ester (1-monolinolenin) a | C21H35O3 | 370.295 | [M + NH4]+ | 353, 261, 243 | 7.1 × 106 | 0.11 |
| 353.269 | [M + H]+ | 261, 243 | ||||||
| Fatty acid amides (Cluster J) | ||||||||
| 51 | 22.82 | 9,12-octadecadienamide b | C18H33NO | 280.264 | [M + H]+ | 263, 245 | 1.5 × 106 | 0.02 |
| 52 | 24.80 | 9-octadecenamide b | C18H35NO | 282.279 | [M + H]+ | 265, 247 | 5.6 × 107 | 0.86 |
| 55 | 27.12 | 9-eicosenoic acid formamide b | C20H39NO | 310.310 | [M + H]+ | 293, 275 | 4.2 × 106 | 0.06 |
| 60 | 29.11 | 13-docosenamide b | C22H43NO | 338.342 | [M + H]+ | 321, 303 | 3.6 × 107 | 0.55 |
| Chlorophyll derivatives (Cluster K) | ||||||||
| 53 | 25.88 | Pheophorbide-a a* | C35H36N4O5 | 593.277 | [M + H]+ | 533, 459, 447 | 2.0 × 107 | 0.31 |
| 56 | 27.98 | Pheophorbide-a methyl ester c | C36H38N4O5 | 607.292 | [M + H]+ | 575, 487, 447 | 2.1 × 107 | 0.32 |
| Carotenoids (Cluster L) | ||||||||
| 54 | 26.59 | Violaxanthin b | C40H56O4 | 601.425 | [M + H]+ | 583, 521, 221, 145, 119 | 2.6 × 106 | 0.04 |
| 57 | 28.00 | β-apo-12′-luteinal c | C25H34O2 | 367.263 | [M + H]+ | 349, 161, 145, 119 | 1.2 × 106 | 0.02 |
| 58 | 28.00 | Antheraxanthin b | C40H56O3 | 585.430 | [M + H]+ | 567, 549, 221, 145, 119 | 3.8 × 107 | 0.58 |
| 59 | 28.02 | Lutein b | C40H56O2 | 568.428 | [M]●+ | 430, 145, 135, 119 | 1.8 × 108 | 2.76 |
| Miscellaneous compounds (“self-loop” nodes) | ||||||||
| Organic acids | ||||||||
| 1 | 0.90 | D-Gluconic acid b | C6H12O7 | 195.050 | [M − H]− | 177, 147, 129, 111 | 3.2 × 108 | 4.91 |
| 4 | 1.18 | Citric acid b | C6H8O7 | 191.019 | [M − H]− | 173, 129, 111 | 8.3 × 106 | 0.13 |
| Nucleoside | ||||||||
| 3 | 1.18 | Adenosine b | C10H13N5O4 | 268.104 | [M + H]+ | 136, 119 | 4.0 × 107 | 0.61 |
| Amino acids | ||||||||
| 5 | 1.21 | Tyrosine b | C9H11NO3 | 182.081 | [M + H]+ | 165, 136, 119 | 6.8 × 106 | 0.10 |
| 8 | 2.19 | Phenylalanine b | C9H11NO2 | 166.086 | [M + H]+ | 120, 103 | 2.1 × 107 | 0.32 |
| 12 | 3.21 | Tryptophan b | C11H12N2O2 | 205.097 | [M + H]+ | 188, 159 | 1.8 × 107 | 0.28 |
| Position | δH, Mult. (J in Hz) | δC | HMBC | δC * | δC ** | |
|---|---|---|---|---|---|---|
| 2J | 3J | |||||
| Apigenin aglycone | ||||||
| 2 | 163.57 | 163.32 | ||||
| 3 | 6.62 s | 102.93 | C-4, C-2 | C-10, C-1′ | 102.60 | |
| 4 | 182.10 | 181.73 | ||||
| 5 | 160.62 | 160.64 | ||||
| 6 | 108.43 | 108.95 | ||||
| 7 | 167.48 | 163.32 | ||||
| 8 | 6.44 s | 94.67 | C-7, C-9 | C-10, C-6 | 93.79 | |
| 9 | 157.57 | 156.31 | ||||
| 10 | 102.57 | 102.95 | ||||
| 1′ | 124.37 | 121.00 | ||||
| 2′ | 7.93 d (8.9) | 127.57 | C-3′ | C-6′, C-4′, C-2 | 128.35 | |
| 3′ | 7.20 d (8.9) | 116.37 | C-4′ | C-5′, C-1′ | 116.01 | |
| 4′ | 160.17 | 161.32 | ||||
| 5′ | 7.20 d (8.9) | 116.37 | C-4′ | C-3′, C-1′ | 116.01 | |
| 6′ | 7.93 d (8.9) | 127.57 | C-5′ | C-4′, C-2′, C-2 | 128.35 | |
| 6-C-β-glucoside | ||||||
| 1″ | 4.91 d (9.9) | 73.91 | C-6, C-2″ | C-5, C-7, C-3″, C-5″ | 73.13 | |
| 2″ | 4.27 m | 70.80 | C-1″, C-3″ | 70.52 | ||
| 3″ | 3.49 t (8.9) | 78.85 | C-2″ | C-5″, C-1″ | 78.93 | |
| 4″ | 3.53 m | 70.09 | C-3″, C-5″ | C-6″ | 70.20 | |
| 5″ | 3.42 m | 80.97 | C-4″ | 81.38 | ||
| 6″ a | 3.87 dd (12.0, 2.0) | 61.18 | C-5″ | C-4″ | 61.37 | |
| 6″ b | 3.76 dd (12.0, 5.0) | |||||
| 4′-O-α-apiofuranoside | ||||||
| 1‴ | 5.69 d (3.0) | 106.96 | C-2‴ | C-4′, C-4‴, C-3‴ | 109.10 | |
| 2‴ | 4.29 d (3.0) | 76.83 | C-1‴ | C-5‴ | 75.90 | |
| 3‴ | 78.94 | 78.70 | ||||
| 4‴ a | 4.14 d (9.8) | 74.26 | C-3‴ | C-1‴, C-5‴, C-2‴ | 73.30 | |
| 4‴ b | 3.92 d (9.8) | |||||
| 5‴ a | 3.65 d (7.3) | 63.05 | C-3‴ | C-2‴, C-4‴ | 63.30 | |
| 5‴ b | 3.65 d (7.3) | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norazhar, A.I.; Lee, S.Y.; Faudzi, S.M.M.; Shaari, K. Metabolite Profiling of Christia vespertilionis Leaf Metabolome via Molecular Network Approach. Appl. Sci. 2021, 11, 3526. https://doi.org/10.3390/app11083526
Norazhar AI, Lee SY, Faudzi SMM, Shaari K. Metabolite Profiling of Christia vespertilionis Leaf Metabolome via Molecular Network Approach. Applied Sciences. 2021; 11(8):3526. https://doi.org/10.3390/app11083526
Chicago/Turabian StyleNorazhar, Anis Irfan, Soo Yee Lee, Siti Munirah Mohd Faudzi, and Khozirah Shaari. 2021. "Metabolite Profiling of Christia vespertilionis Leaf Metabolome via Molecular Network Approach" Applied Sciences 11, no. 8: 3526. https://doi.org/10.3390/app11083526