Chemotherapy-Induced Survivin Regulation in Acute Myeloid Leukemia Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Cell Counting
2.3. Flow-Cytometry
2.4. Real-Time PCR
2.5. Immunoblotting
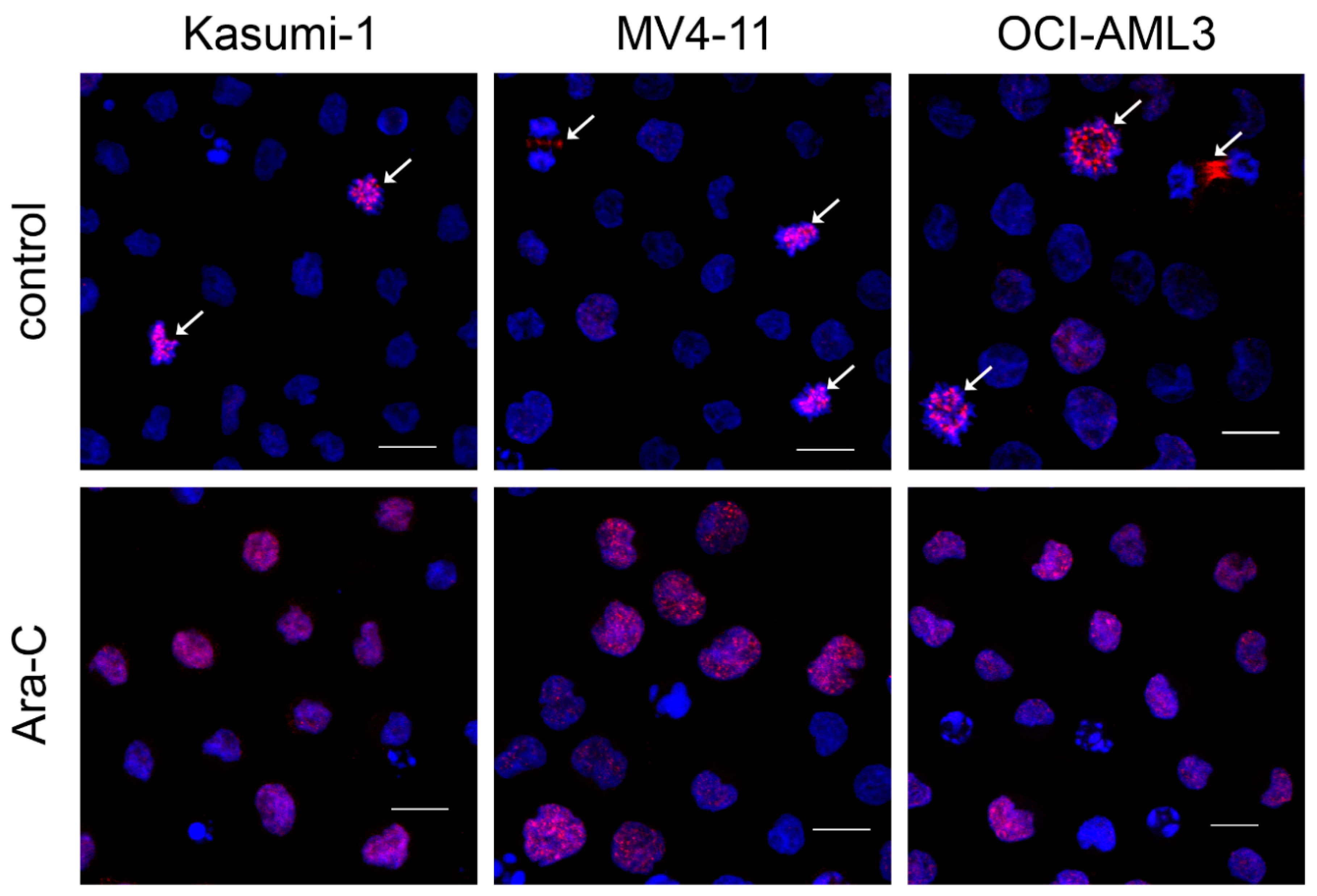
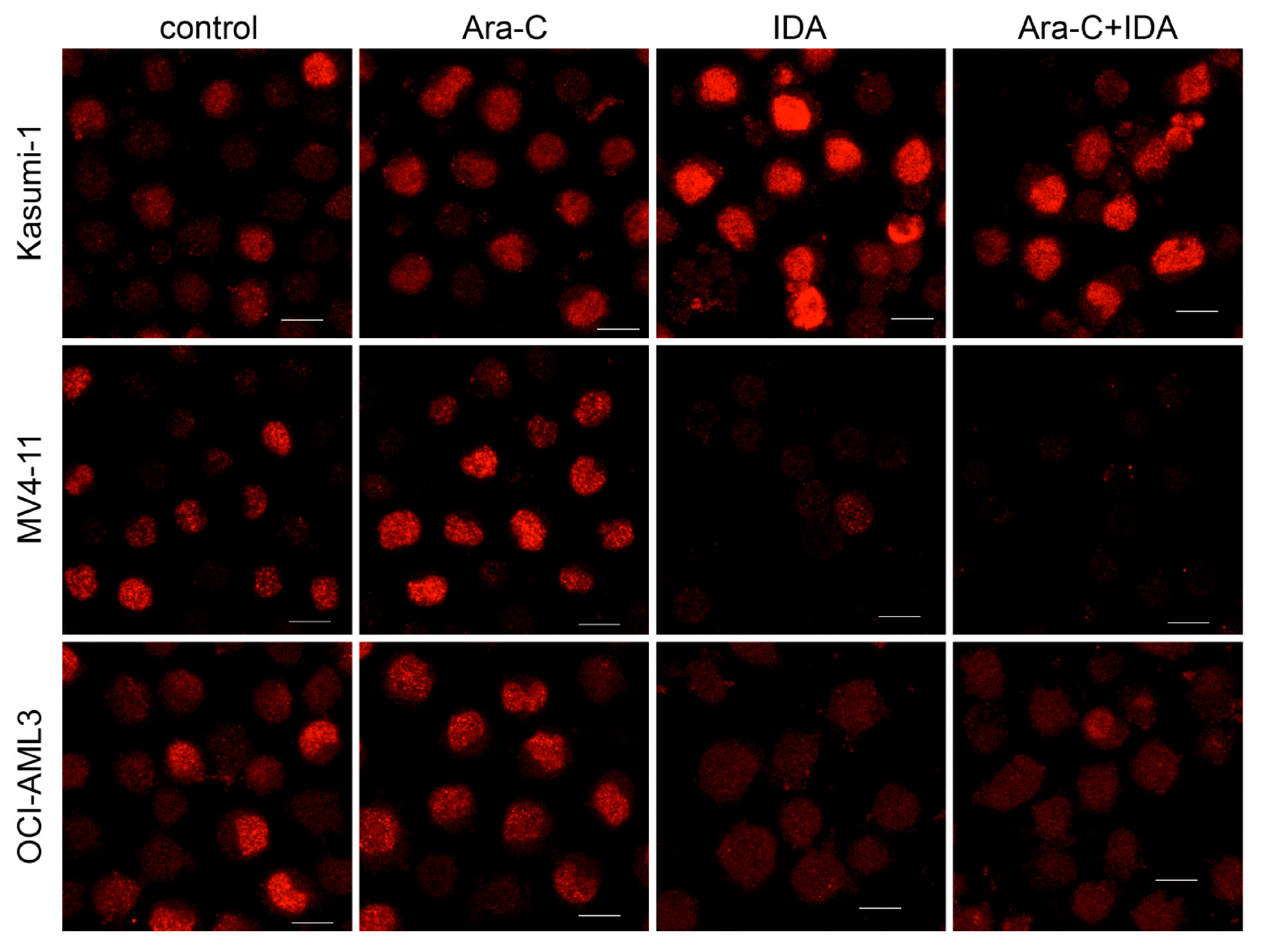
2.6. Immunofluorescence
2.7. Statistical Analyses
3. Results
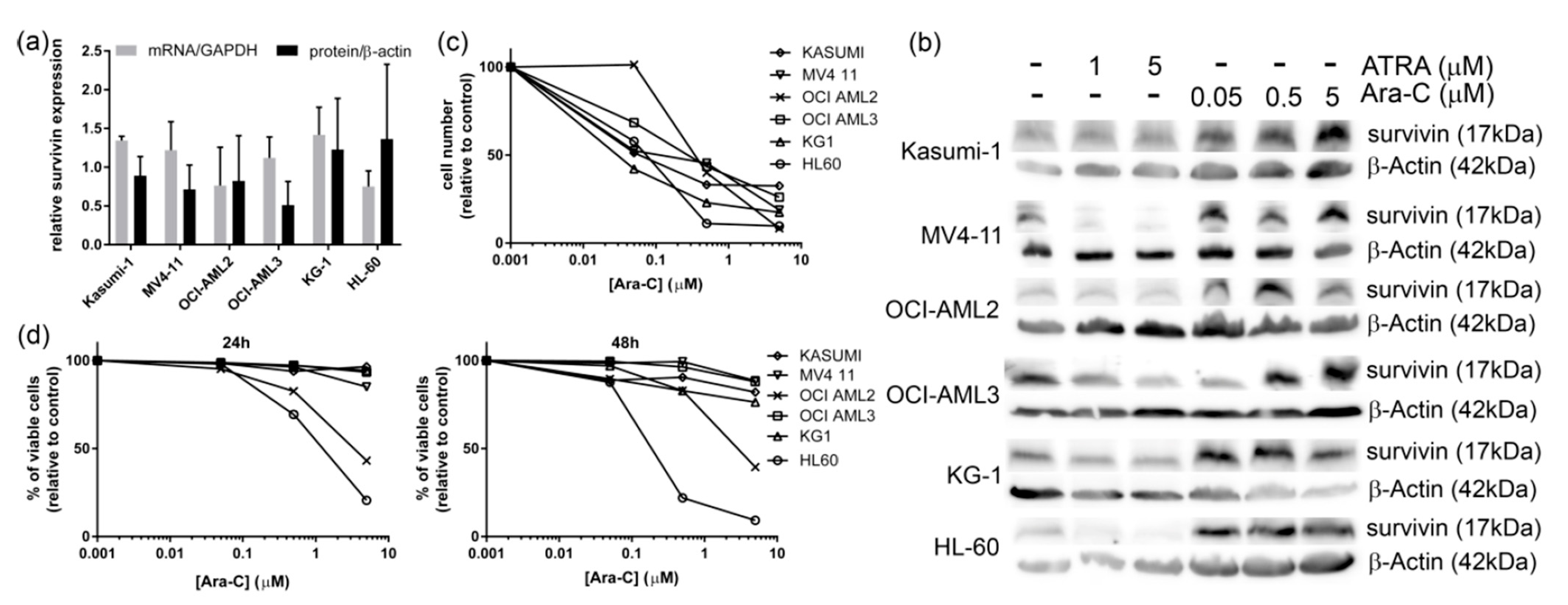
3.1. Ara-C Treatment
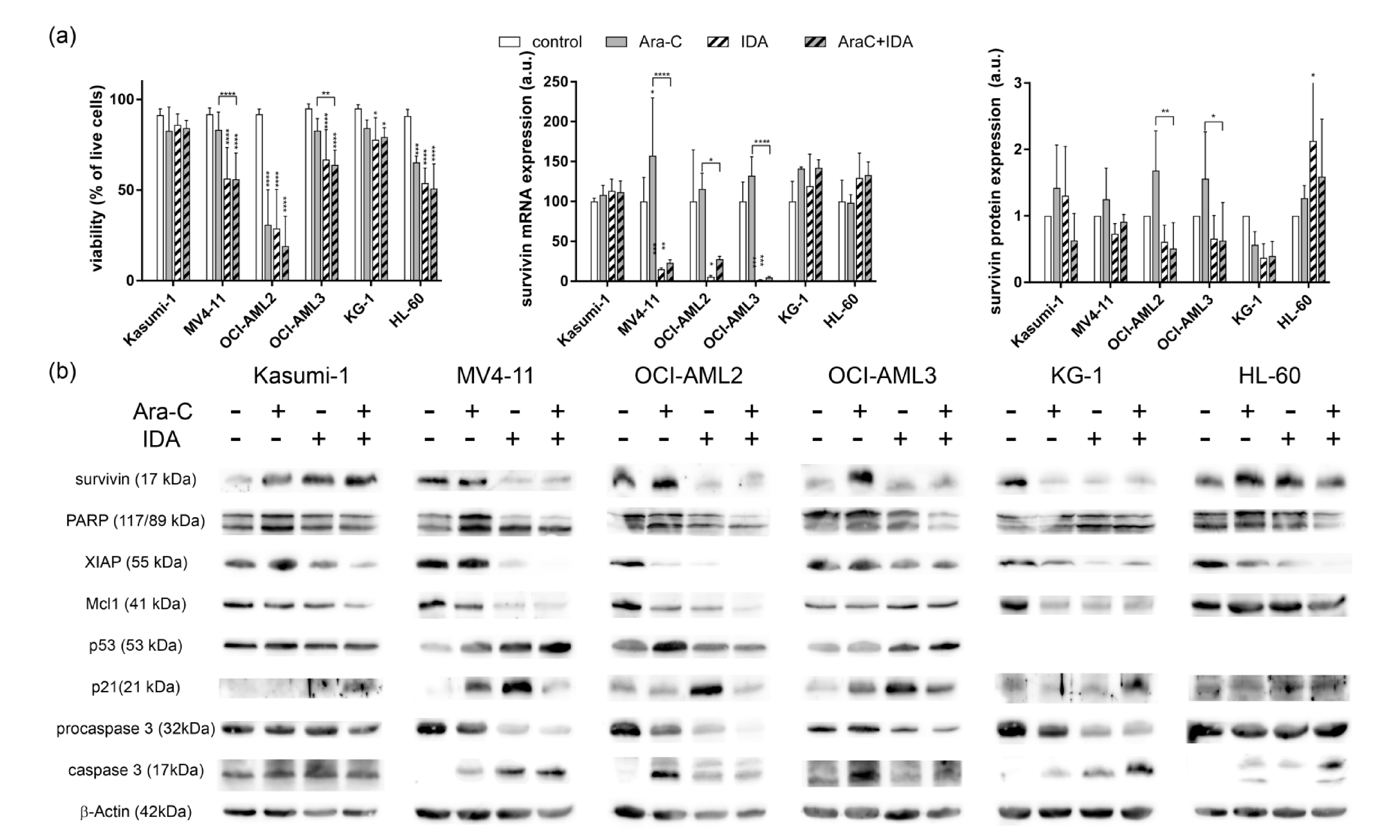
3.2. Combination with IDA
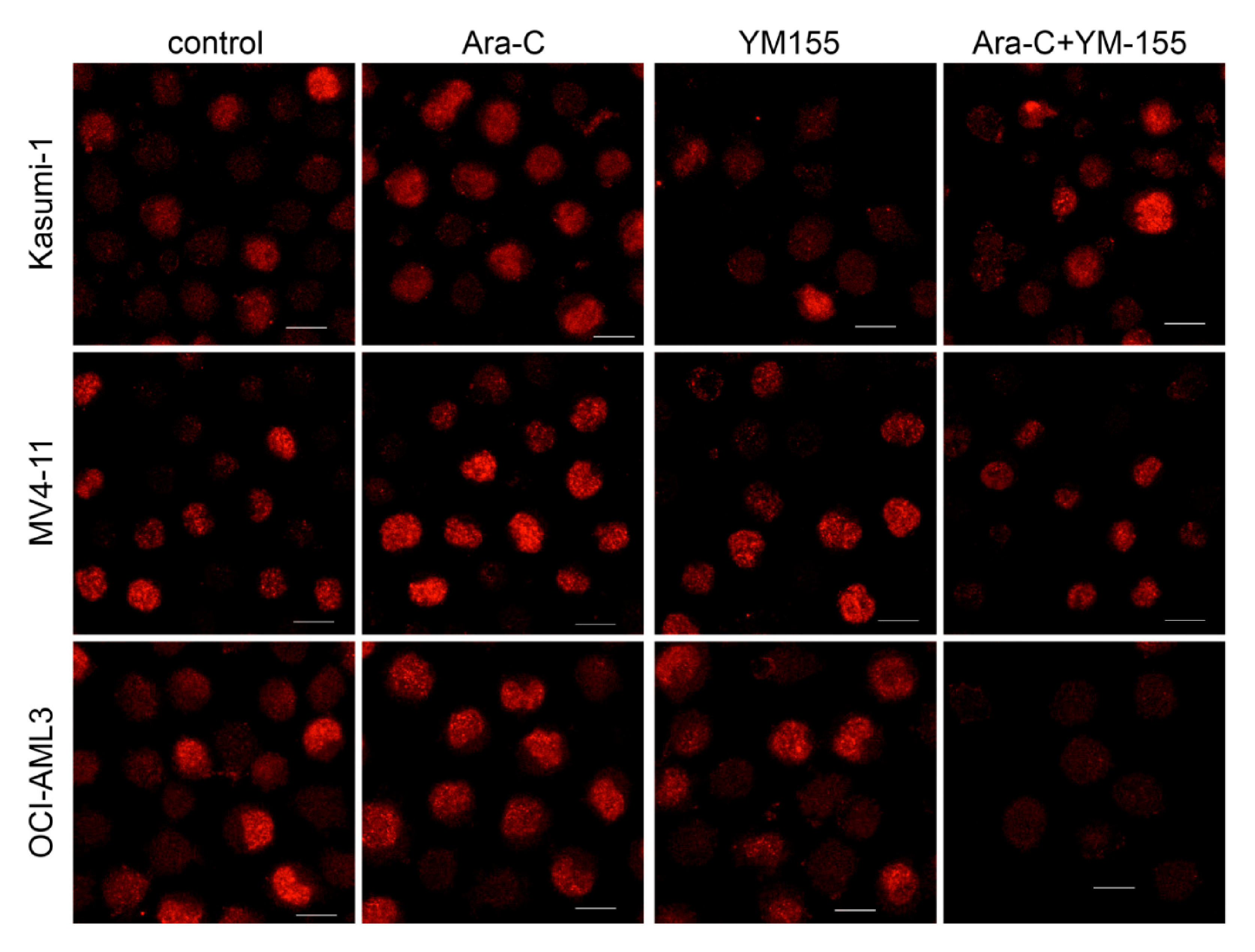
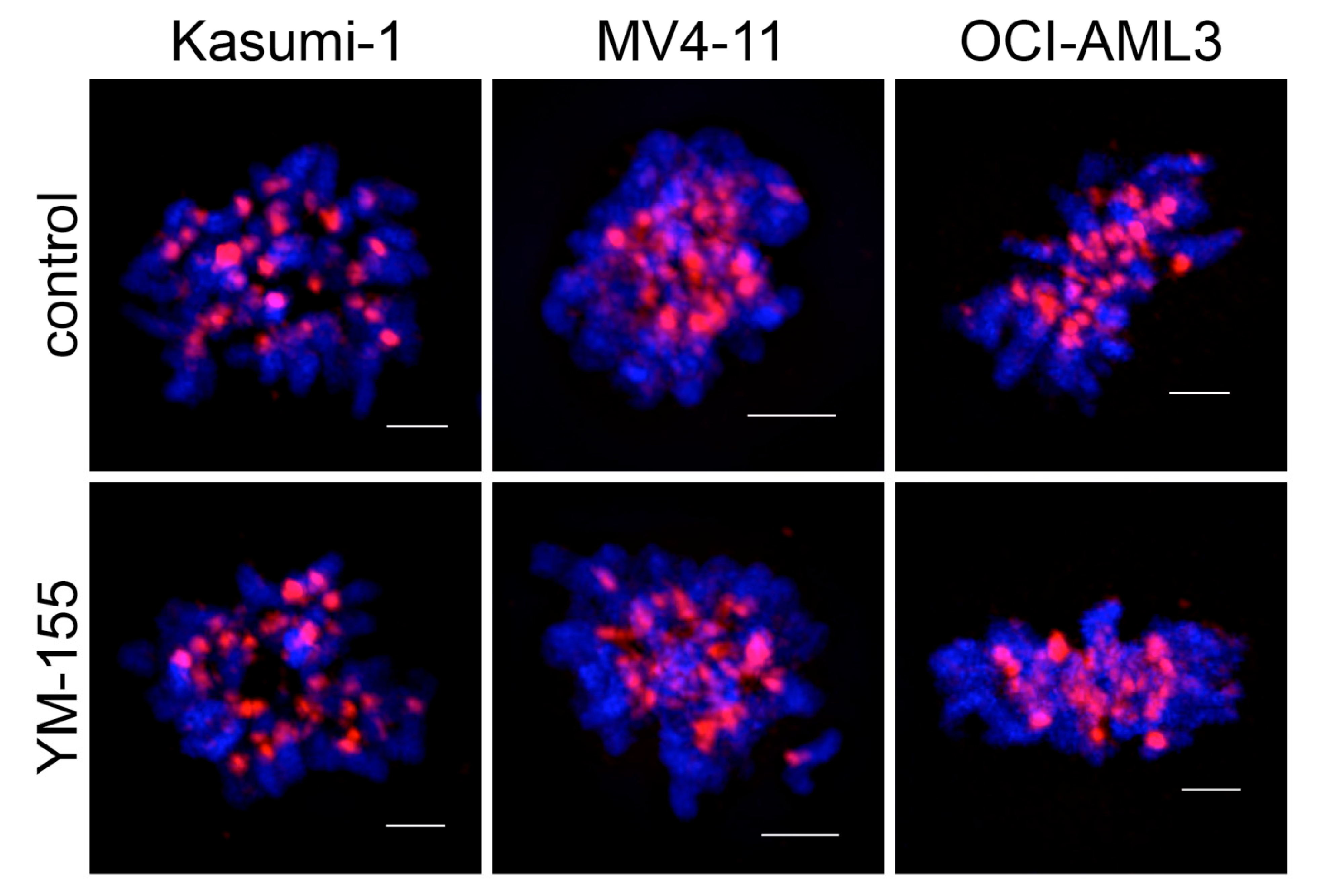
3.3. Survivin Inhibition with YM-155
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Saltarella, I.; Vinella, A.; Muzio, L.L.; Pannone, G.; Fumarulo, R.; Vacca, A.; Mariggiò, M.A. Survivin overexpression in head and neck squamous cell carcinomas as a new therapeutic target (Review). Oncol. Rep. 2019, 41, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yang, K.; Wang, H.; Chen, X.; Wu, H.; Yao, L.; Ma, S. Expression and clinical significance of survivin in ovarian cancer: A meta-analysis. PLoS ONE 2018, 13, e0194463. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Gao, W.; Kang, Q.M.; Zhang, X.J.; Yang, S.G. Prognostic value of survivin in patients with gastric cancer: A systematic review with meta-analysis. PLoS ONE 2013, 8, e71930. [Google Scholar] [CrossRef] [Green Version]
- Small, S.; Keerthivasan, G.; Huang, Z.; Gurbuxani, S.; Crispino, J.D. Overexpression of survivin initiates hematologic malignancies in vivo. Leukemia 2010, 24, 1920–1926. [Google Scholar] [CrossRef]
- Carter, B.Z.; Milella, M.; Altieri, D.C.; Andreeff, M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood 2001, 97, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Hu, C.; Li, H. Survivin as a novel target protein for reducing the proliferation of cancer cells. Biomed. Rep. 2018, 8, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, S.P. The functional repertoire of survivin’s tails. Cell Cycle 2015, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Angell, H. A study into the potential role of Survivin localization in resistance to drug-induced apoptosis. Biosci. Horiz. 2008, 1, 85–91. Available online: https://academic.oup.com/biohorizons/article/1/2/85/269002 (accessed on 26 May 2015). [CrossRef] [Green Version]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Donahue, J.; Smith, A.; Hornick, J.; Rao, J.N.; Wang, J.; Battafarano, R.J. Loss of p53, rather than beta-catenin overexpression, induces survivin-mediated resistance to apoptosis in an esophageal cancer cell line. J. Thorac. Cardiovasc. Surg. 2010, 140, 225–232. [Google Scholar] [CrossRef] [Green Version]
- De Cesare, M.; Cominetti, D.; Doldi, V.; Lopergolo, A.; Deraco, M.; Gandellini, P.; Friedlander, S.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; et al. Anti-tumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: The role of survivin. Oncotarget 2015, 6, 13119–13132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Hennig, D.; Schäfer, C.; Wirth, M.; Marx, C.; Heinzel, T.; Schneider, G.; Krämer, O.H. Survivin and YM155: How faithful is the liaison? Biochim. Biophys. Acta 2014, 1845, 202–220. [Google Scholar] [CrossRef]
- Véquaud, E.; Desplanques, G.; Jézéquel, P.; Juin, P.; Barillé-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Hu, L.; Li, Y.; Dou, L.; Cai, P.; Tang, Z.; Wang, L. Effect of survivin regulation of transcription level by p21waf1 overexpression in HepG2 hepatocellular carcinoma cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2008, 28, 308–313. [Google Scholar] [CrossRef]
- Suzuki, A.; Ito, T.; Kawano, H.; Hayashida, M.; Hayasaki, Y.; Tsutomi, Y.; Akahane, K.; Nakano, T.; Miura, M.; Shiraki, K. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene 2000, 19, 1346–1353. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Carlstedt, A.; Emmerich, C.; Mustafa, A.M.; Göder, A.; Knauer, S.K.; Linnebacher, M.; Heinzel, T.; Krämer, O.H. Survivin antagonizes chemotherapy-induced cell death of colorectal cancer cells. Oncotarget 2018, 9, 27835–27850. [Google Scholar] [CrossRef]
- Fukuda, S.; Abe, M.; Onishi, C.; Taketani, T.; Purevsuren, J.; Yamaguchi, S.; Conway, E.M.; Pelus, L.M. Survivin selectively modulates genes deregulated in human leukemia stem cells. J. Oncol. 2011, 2011, 946936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Little, E.B.; Zivanovic, A.; Hong, P.; Liu, A.K.S.; Burow, R.; Stinson, C.; Hallahan, A.R.; Moore, A.S. Targeting survivin with YM155 (Sepantronium Bromide): A novel therapeutic strategy for paediatric acute myeloid leukaemia. Leuk. Res. 2015, 39, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Wheat, J.; Chen, X.; Jin, S.; Sadrzadeh, H.; Fathi, A.T.; Peterson, R.T.; Kung, A.L.; Sweetser, D.A.; et al. AML1-ETO mediates hematopoietic self-renewal and leukemogenesis through a COX/β-catenin signaling pathway. Blood 2013, 121, 4906–4916. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Ookura, M.; Zokumasu, K.; Ueda, T. Gö6976, a FLT3 kinase inhibitor, exerts potent cytotoxic activity against acute leukemia via inhibition of survivin and MCL-1. Biochem. Pharmacol. 2014, 90, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. New wirings in the survivin networks. Oncogene 2008, 27, 6276–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Liu, B.; Eklund, E.A. Investigating the role of the innate immune response in relapse or blast crisis in chronic myeloid leukemia. Leukemia 2020. [Google Scholar] [CrossRef] [Green Version]
- Wiedemuth, R.; Klink, B.; Töpfer, K.; Schröck, E.; Schackert, G.; Tatsuka, M.; Temme, A. Survivin safeguards chromosome numbers and protects from aneuploidy independently from p53. Mol. Cancer 2014, 13, 107. [Google Scholar] [CrossRef] [Green Version]
- Mera, S.; Magnusson, M.; Tarkowski, A.; Bokarewa, M. Extracellular survivin up-regulates adhesion molecules on the surface of leukocytes changing their reactivity pattern. J. Leukoc. Biol. 2008, 83, 149–155. [Google Scholar] [CrossRef]
- Niedbala, W.; Cai, B.; Liu, H.; Pitman, N.; Chang, L.; Liew, F.Y. Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL-2, and OX40. Proc. Natl. Acad. Sci. USA 2007, 104, 15478–15483. [Google Scholar] [CrossRef] [Green Version]
- Knutsen, A.; Adell, G.; Sun, X. Survivin expression is an independent prognostic factor in rectal cancer patients with and without preoperative radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 149–155. [Google Scholar] [CrossRef]
- Tong, X.; Yang, P.; Wang, K.; Liu, Y.; Liu, X.; Shan, X.; Huang, R.; Zhang, K.; Wang, J. Survivin is a prognostic indicator in glioblastoma and may be a target of microRNA-218. Oncol. Lett. 2019, 18, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, H.; Li, R.; Guo, Y.; Zheng, R. High expression of survivin predicts poor prognosis in cervical squamous cell carcinoma treated with paclitaxel and carboplatin. Medicine 2019, 98, e15607. [Google Scholar] [CrossRef] [PubMed]
- Stroopinsky, D.; Rajabi, H.; Nahas, M.; Rosenblatt, J.; Rahimian, M.; Pyzer, A.; Tagde, A.; Kharbanda, A.; Jain, S.; Kufe, T.; et al. MUC1-C drives myeloid leukaemogenesis and resistance to treatment by a survivin-mediated mechanism. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Schmelz, K.; Wuchter, C.; Ludwig, W.; Dörken, B.; Tamm, I. In vivo expression of survivin and its splice variant survivin-2B: Impact on clinical outcome in acute myeloid leukemia. Int. J. Cancer 2006, 119, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Balkhi, M.Y.; Christopeit, M.; Chen, Y.; Geletu, M.; Behre, G. AML1/ETO-induced survivin expression inhibits transcriptional regulation of myeloid differentiation. Exp. Hematol. 2008, 36, 1449–1460. [Google Scholar] [CrossRef]
- Morrison, D.J.; Hogan, L.E.; Condos, G.; Bhatla, T.; Germino, N.; Moskowitz, N.P.; Lee, L.; Bhojwani, D.; Horton, T.M.; Belitskaya-Levy, I.; et al. Endogenous knockdown of survivin improves chemotherapeutic response in ALL models. Leukemia 2012, 26, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Daglioglu, C.; Kaci, F.N. Cascade therapy with doxorubicin and survivin-targeted tailored nanoparticles: An effective alternative for sensitization of cancer cells to chemotherapy. Int. J. Pharm. 2019, 561, 74–81. [Google Scholar] [CrossRef]
- Nestal de Moraes, G.; Silva, K.L.; Vasconcelos, F.d.C.; Maia, R.C. Survivin overexpression correlates with an apoptosis-resistant phenotype in chronic myeloid leukemia cells. Oncol. Rep. 2011, 25, 1613–1619. [Google Scholar] [CrossRef] [Green Version]
- De Souza Reis, R.R.; Casal de Faria, F.C.; Castro, C.P.; de Souza, P.S.; da Cunha Vasconcelos, F.; Bello, R.D.; da Silva, A.J.; Costa, P.R.R.; Maia, R.C. The therapeutical potential of a novel pterocarpanquinone LQB-118 to target inhibitor of apoptosis proteins in acute myeloid leukemia cells. Anticancer Agents Med. Chem. 2013, 13, 341–351. [Google Scholar] [CrossRef]
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Hatakeyama, S.; Kinoyama, I.; Matsuhisa, A.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Kudchadkar, R.; Ernst, S.; Chmielowski, B.; Redman, B.G.; Steinberg, J.; Keating, A.; Jie, F.; Chen, C.; Gonzalez, R.; Weber, J. A phase 2, multicenter, open-label study of sepantronium bromide (YM155) plus docetaxel in patients with stage III (unresectable) or stage IV melanoma. Cancer Med. 2015, 4, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Danielpour, D.; Gao, Z.; Zmina, P.M.; Shankar, E.; Shultes, B.C.; Jobava, R.; Welford, S.M.; Hatzoglou, M. Early Cellular Responses of Prostate Carcinoma Cells to Sepantronium Bromide (YM155) Involve Suppression of mTORC1 by AMPK. Sci. Rep. 2019, 9, 11541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Nakahara, T.; Yamauchi, T.; Kita, A.; Takeuchi, M.; Kiyonaga, F.; Kaneko, N.; Sasamata, M. Antitumor activity of YM155, a selective small-molecule survivin suppressant, alone and in combination with docetaxel in human malignant melanoma models. Clin. Cancer Res. 2011, 17, 5423–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccone, G.; Zatloukal, P.; Roubec, J.; Floor, K.; Musil, J.; Kuta, M.; van Klaveren, R.J.; Chaudhary, S.; Gunther, A.; Shamsili, S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 4481–4486. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Yoshida, A.; Ueda, T. YM155 induces caspase-8 dependent apoptosis through downregulation of survivin and Mcl-1 in human leukemia cells. Biochem. Biophys. Res. Commun. 2013, 435, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.H.; Johnson, K.; LaTocha, D.; Rowley, J.S.J.; Bryant, J.; Burke, R.; Smith, R.L.; Loriaux, M.; Müschen, M.; Mullighan, C.; et al. YM155 potently kills acute lymphoblastic leukemia cells through activation of the DNA damage pathway. J. Hematol. Oncol. 2015, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Brodska, B.; Holoubek, A.; Otevrelova, P.; Kuzelova, K. Low-Dose Actinomycin-D Induces Redistribution of Wild-Type and Mutated Nucleophosmin Followed by Cell Death in Leukemic Cells. J. Cell Biochem. 2016, 117, 1319–1329. [Google Scholar] [CrossRef]
- Brodska, B.; Otevrelova, P.; Holoubek, A. Decitabine and SAHA-induced apoptosis is accompanied by survivin downregulation and potentiated by ATRA in p53-deficient cells. Oxid. Med. Cell. Longev. 2014, 2014, 165303. [Google Scholar] [CrossRef]
- Van Pelt, K.; de Haan, G.; Vellenga, E.; Daenen, S.M.G.J. Administration of low-dose cytarabine results in immediate S-phase arrest and subsequent activation of cell cycling in murine stem cells. Exp. Hematol. 2005, 33, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, A.; Shao, M.; Lin, L.; Li, P.; Wang, Y. Schisandrin B reverses doxorubicin resistance through inhibiting P-glycoprotein and promoting proteasome-mediated degradation of survivin. Sci. Rep. 2017, 7, 8419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareková, M.; Vávrová, J.; Vokurková, D. Dose dependent biological effects of idarubicin in HL-60 cells: Alterations of the cell-cycle and apoptosis. Acta Medica 2000, 43, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lyu, H.; Wang, J.; Liu, B. Influence of survivin-targeted therapy on chemosensitivity in the treatment of acute myeloid leukemia. Cancer Lett. 2015, 366, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, S.; Singh, P.; Moh, A.; Abe, M.; Conway, E.M.; Boswell, H.S.; Yamaguchi, S.; Fu, X.; Pelus, L.M. Survivin mediates aberrant hematopoietic progenitor cell proliferation and acute leukemia in mice induced by internal tandem duplication of Flt3. Blood 2009, 114, 394–403. [Google Scholar] [CrossRef] [Green Version]
- NSC348884 Cytotoxicity Is Not Mediated by Inhibition of Nucleophosmin Oligomerization. Available online: https://search.datacite.org/works/10.1101/2020.02.06.936666 (accessed on 1 April 2020).
- Colnaghi, R.; Connell, C.M.; Barrett, R.M.A.; Wheatley, S.P. Separating the anti-apoptotic and mitotic roles of survivin. J. Biol. Chem. 2006, 281, 33450–33456. [Google Scholar] [CrossRef] [Green Version]
- Knauer, S.K.; Bier, C.; Habtemichael, N.; Stauber, R.H. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep. 2006, 7, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Erba, H.P.; Sayar, H.; Juckett, M.; Lahn, M.; Andre, V.; Callies, S.; Schmidt, S.; Kadam, S.; Brandt, J.T.; Van Bockstaele, D.; et al. Safety and pharmacokinetics of the antisense oligonucleotide (ASO) LY2181308 as a single-agent or in combination with idarubicin and cytarabine in patients with refractory or relapsed acute myeloid leukemia (AML). Investig. New Drugs 2013, 31, 1023–1034. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otevřelová, P.; Brodská, B. Chemotherapy-Induced Survivin Regulation in Acute Myeloid Leukemia Cells. Appl. Sci. 2021, 11, 460. https://doi.org/10.3390/app11010460
Otevřelová P, Brodská B. Chemotherapy-Induced Survivin Regulation in Acute Myeloid Leukemia Cells. Applied Sciences. 2021; 11(1):460. https://doi.org/10.3390/app11010460
Chicago/Turabian StyleOtevřelová, Petra, and Barbora Brodská. 2021. "Chemotherapy-Induced Survivin Regulation in Acute Myeloid Leukemia Cells" Applied Sciences 11, no. 1: 460. https://doi.org/10.3390/app11010460