Genetic Diversity of Indigenous Pigs from South China Area Revealed by SNP Array

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample and Data
2.3. Genetic Diversity Analysis
2.4. Phylogenetic Analysis
2.5. Population Structure Analysis
2.6. Linkage Disequilibrium and Effective Population Size Analysis
2.7. Genetic Differentiation and Detection of Selection Signatures
2.8. Genome Annotation and Quantitative Trait Loci Overlapping with Potential Selection Signatures
2.9. Enrichment Analysis
3. Results
3.1. Genetic Diversity Analysis
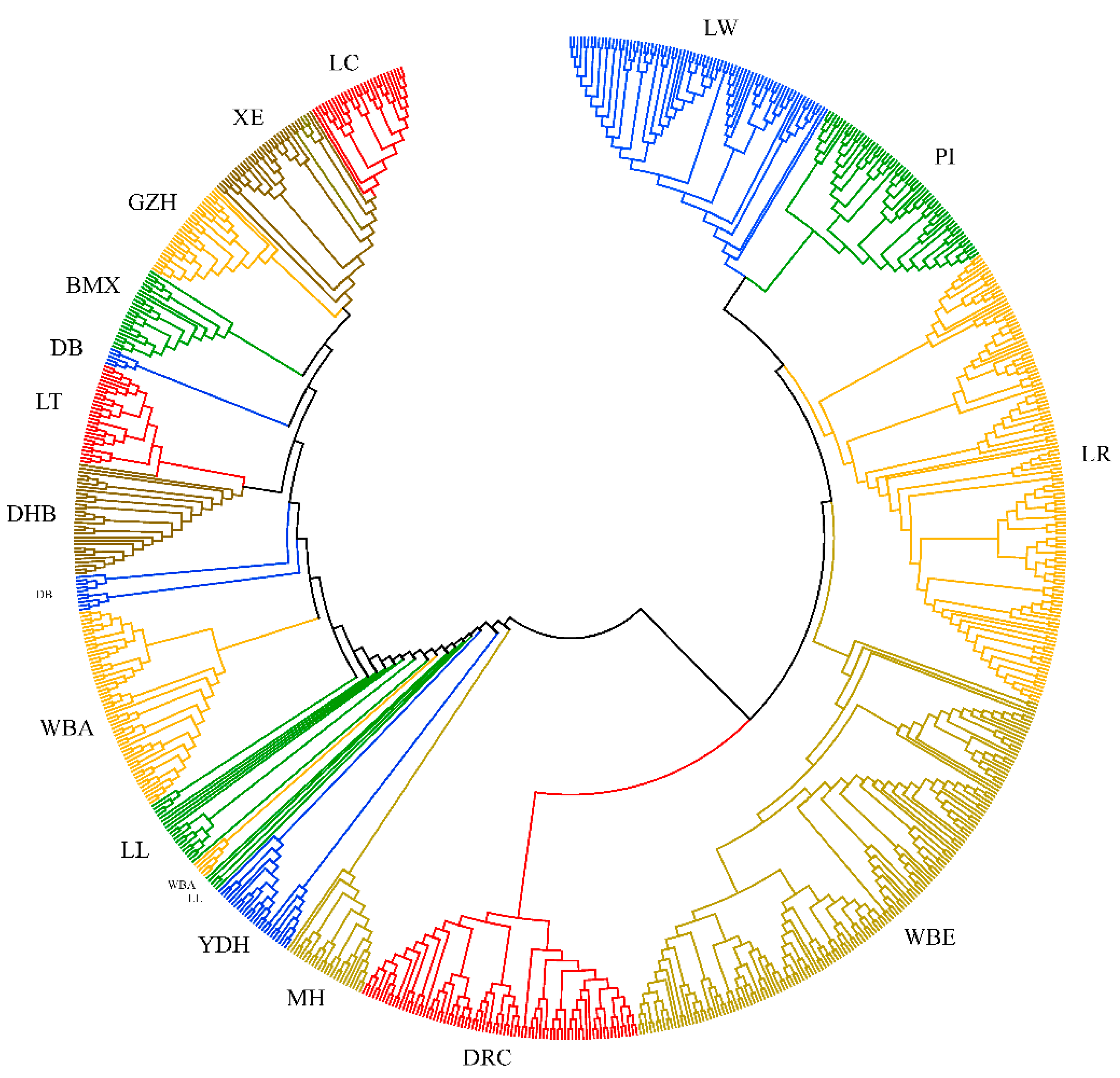
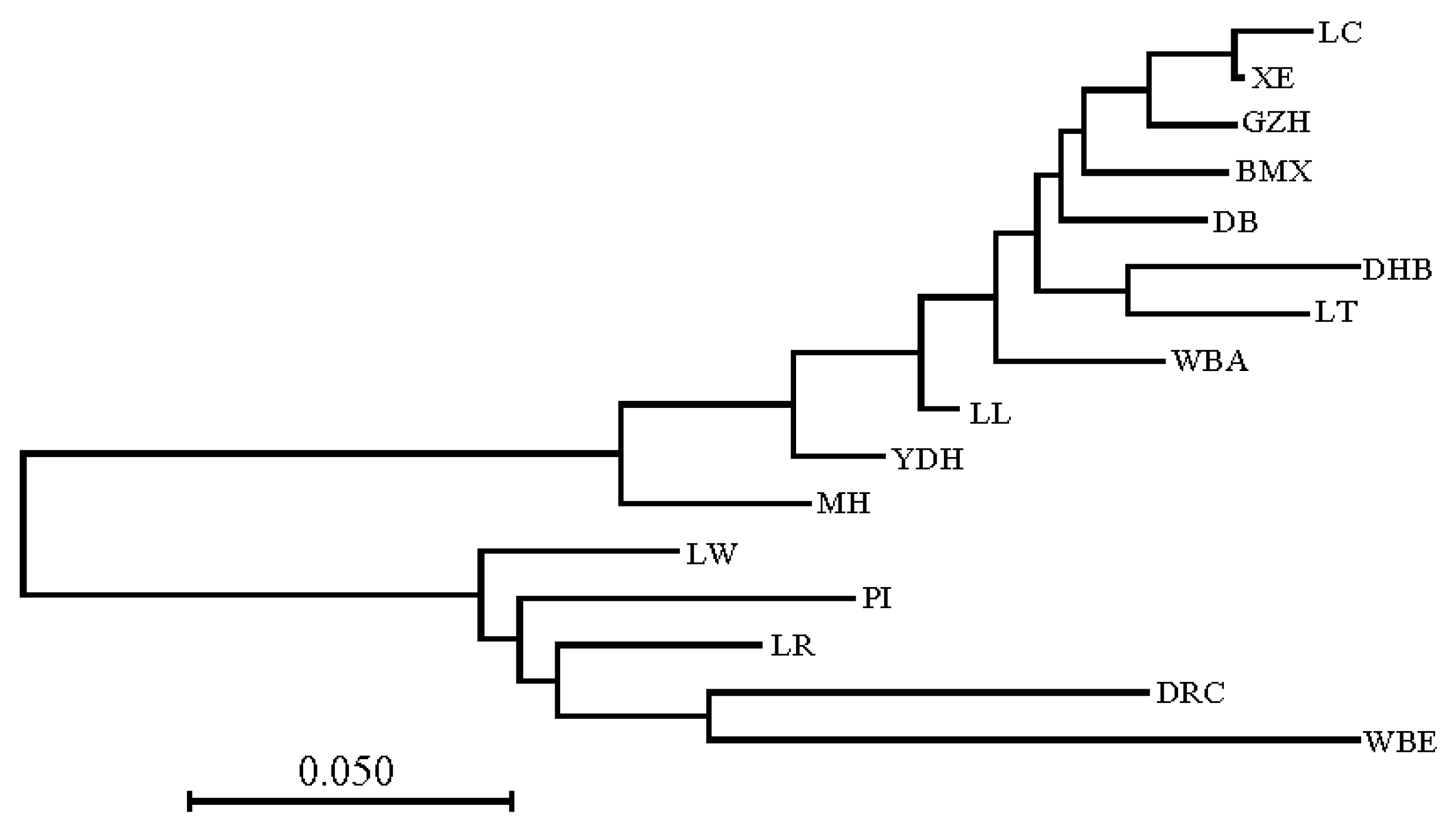
3.2. Genetic Distance Analysis
3.3. Population Structure Analysis
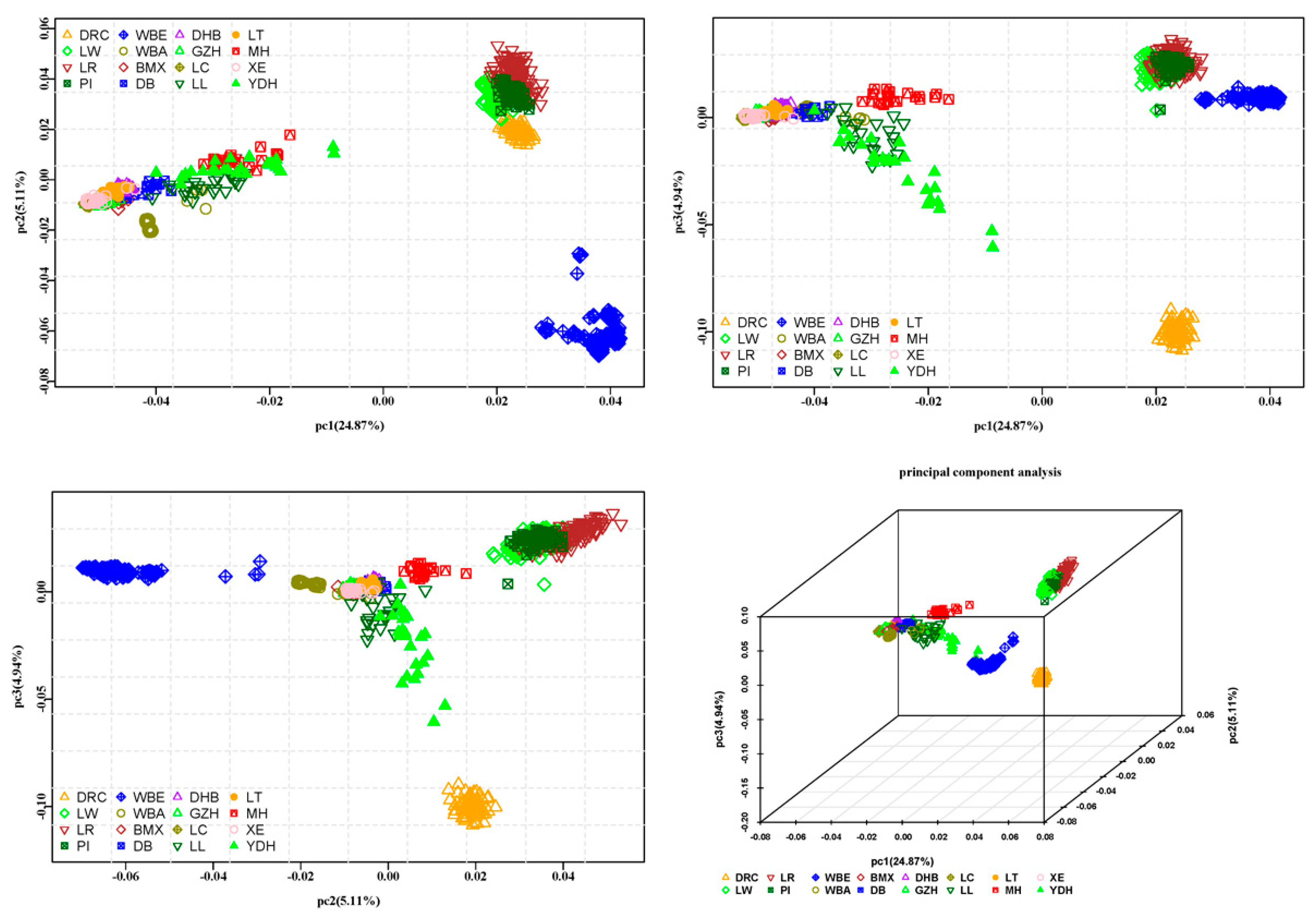
3.3.1. Principal Component Analysis-Based Clustering
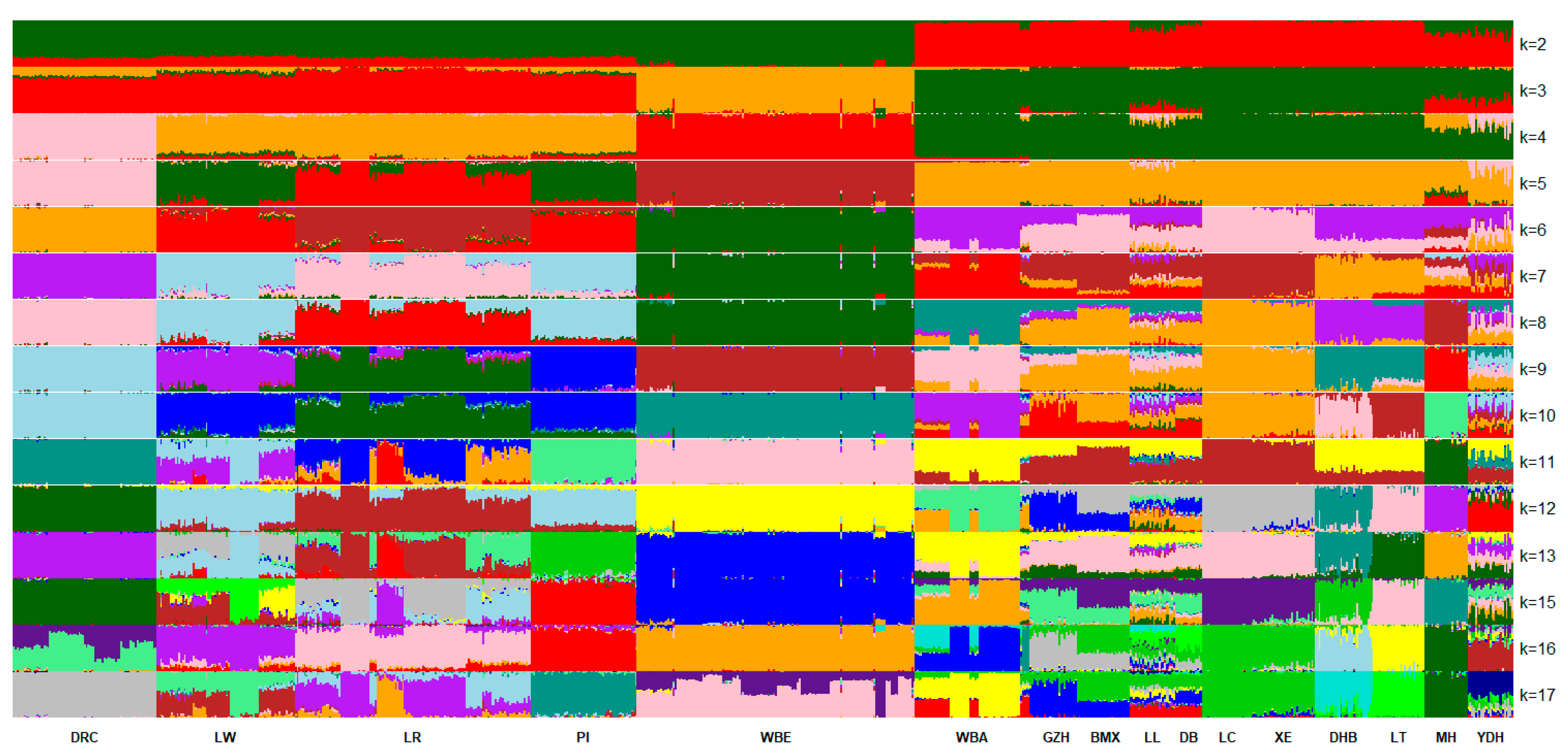
3.3.2. Population Cluster Based on STRUCTURE
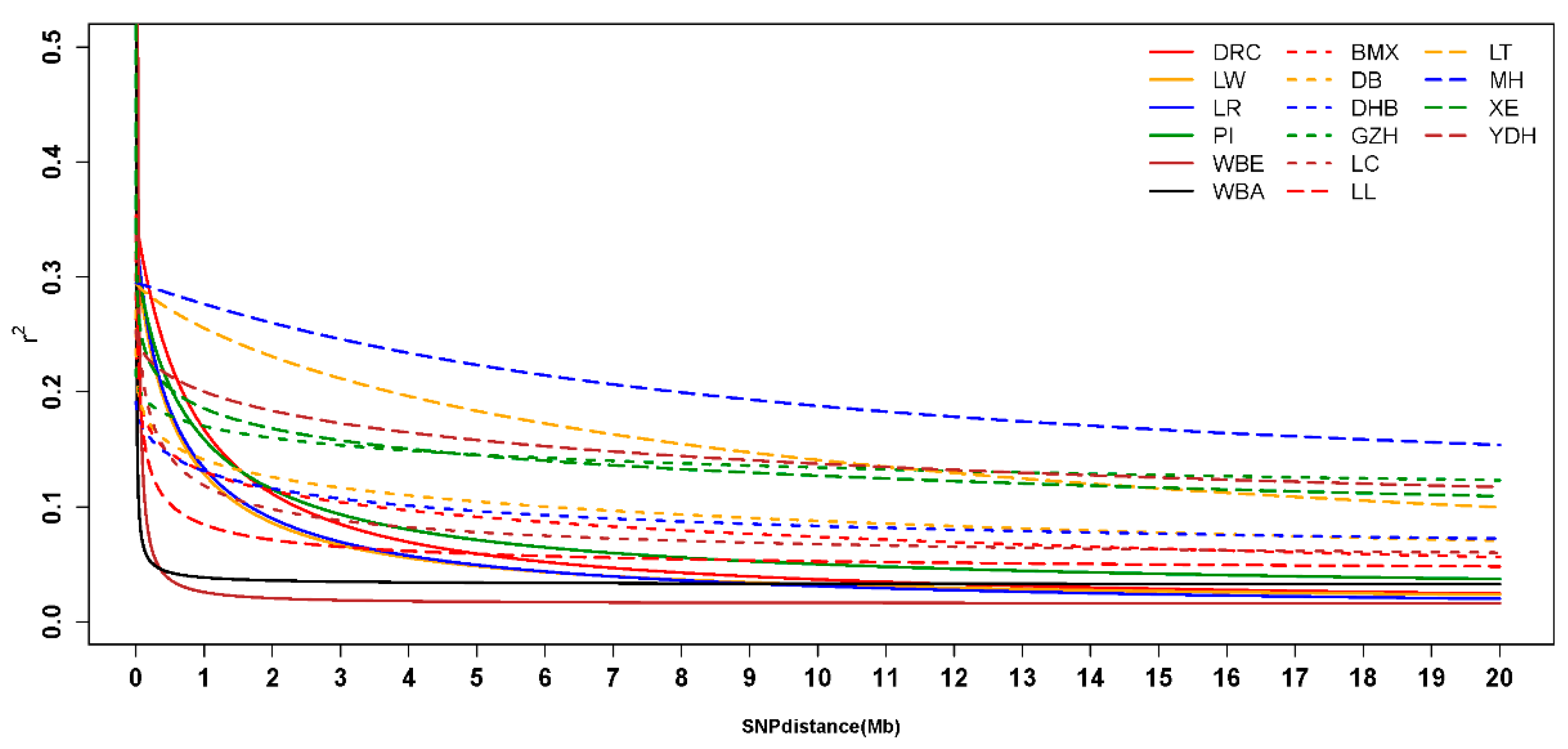
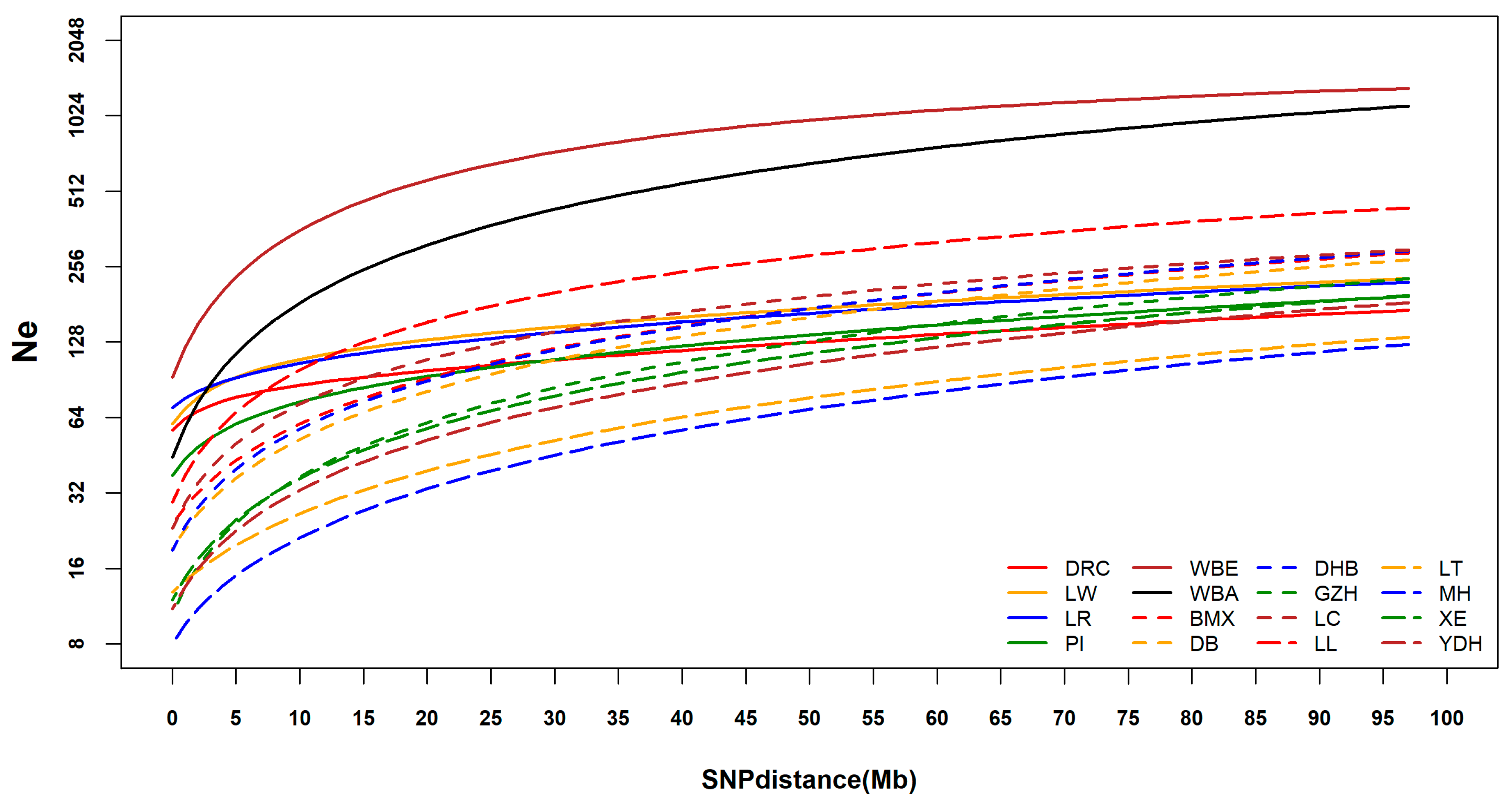
3.4. Linkage Disequilibrium and Analysis of Effective Population Size
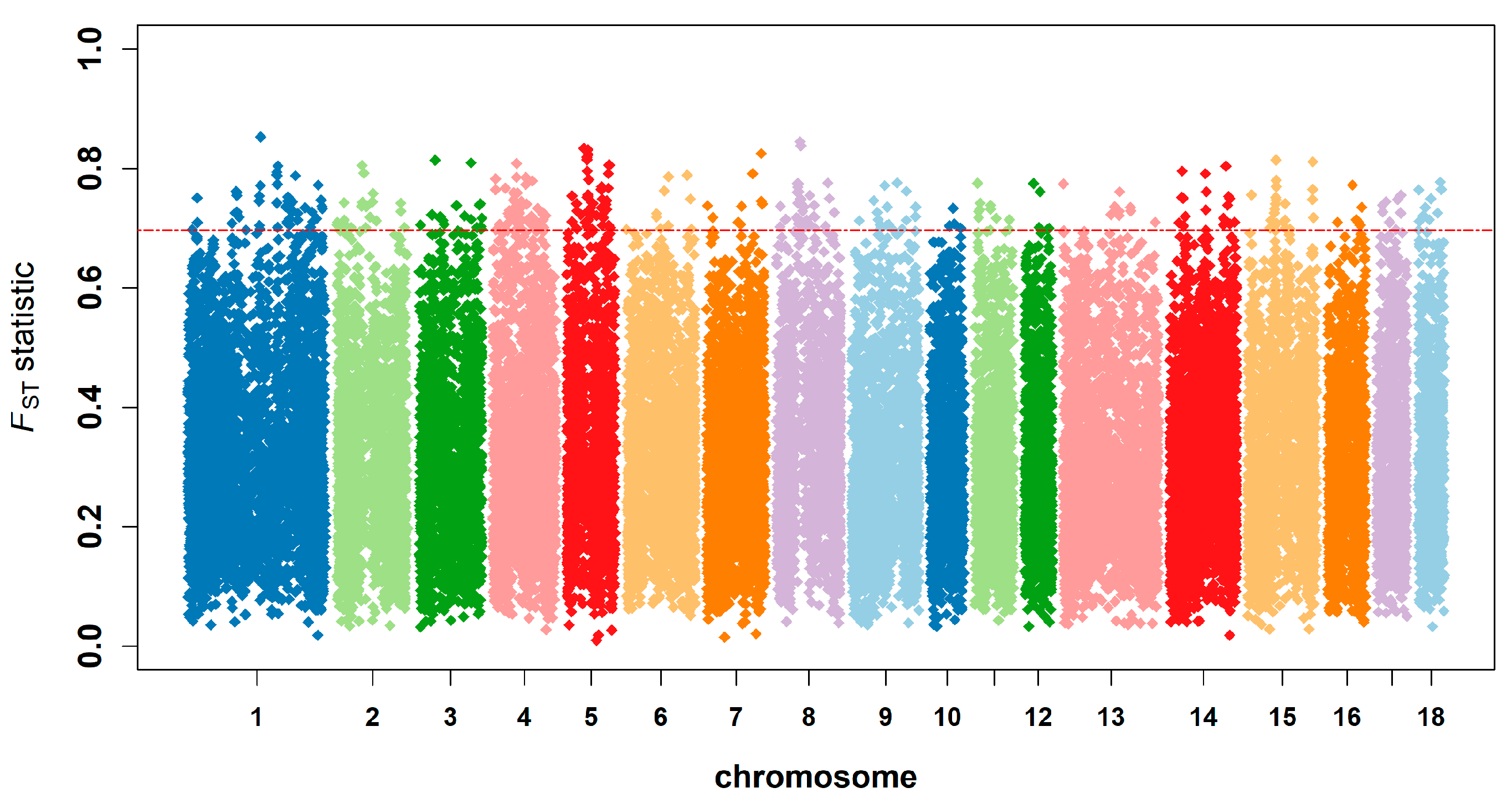
3.5. Genetic Differentiation, Outlier Loci, and Candidate Genes under Selection
3.6. Genome Annotation and Quantitative Trait Loci Overlapping with Potential Selection Signatures
3.7. Enrichment Analysis
4. Discussion
4.1. Genetic Diversity of Indigenous Pigs
4.2. Genetic Distance and Population Structure Analysis
4.3. Linkage Disequilibrium and Effective Population Size Analysis
4.4. Genetic Differentiation and Detection of Selection Signatures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- OECD-FAO Agricultural Outlook. 2018–2027. Available online: http://www.fao.org/publications/oecd-fao-agricultural-outlook/en/ (accessed on 11 June 2019).
- Ai, H.; Fang, X.; Yang, B.; Huang, Z.; Chen, H.; Mao, L.; Zhang, F.; Zhang, L.; Cui, L.; He, W.; et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015, 47, 217–225. [Google Scholar] [CrossRef] [PubMed]
- National Committee of Animal Genetic Resources organization in China. Animal Genetic Resources in China: Pigs; China Agriculture Press: Beijing, China, 2011. [Google Scholar]
- Ai, H.; Huang, L.; Ren, J. Genetic diversity, linkage disequilibrium and selection signatures in chinese and Western pigs revealed by genome-wide SNP markers. PLoS ONE 2013, 8, e56001. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.L. What to Preserve? An Application of Diversity Theory to Crane Conservation? Q. J. Econ. 1993, 108, 157–183. [Google Scholar] [CrossRef]
- Caballero, A.; Toro, M. Interrelations between effective population size and other pedigree tools for the management of conserved populations. Genet. Res. 2000, 75, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Andersson, L. Mitochondrial diversity in European and Chinese pigs is consistent with population expansions that occurred prior to domestication. Proc. Biol. Soi. 2006, 273, 1803–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, B.; Wang, Z.G.; Li, Y.J.; Zhao, X.L.; Liu, B.; Zhao, S.H.; Yu, M.; Li, M.H.; Chen, S.L.; Xiong, T.A.; et al. Genetic variation analysis within and among Chinese indigenous swine populations using microsatellite markers. Anim. Genet. 2002, 33, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Giuffra, E.; Kijas, J.; Amarger, V.; Carlborg, Ö.; Jeon, J.-T.; Andersson, L. The origin of the domestic pig: Independent domestication and subsequent introgression. Genetics 2000, 154, 1785–1791. [Google Scholar]
- Yang, J.; Wang, J.; Kijas, J.; Liu, B.; Li, K. Genetic diversity present within the near-complete mtDNA genome of 17 breeds of indigenous Chinese pigs. J. Hered. 2003, 94, 381–385. [Google Scholar] [CrossRef]
- Chen, M.; Su, G.; Fu, J.; Zhang, Q.; Wang, A.; Lund, M.S.; Guldbrandtsen, B. Population admixture in Chinese and European Sus scrofa. Sci. Rep.-UK 2017, 7. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Wang, Y.; Wu, Y.; Fu, J.; Liu, J.F. Genome-wide detection of selection signatures in Chinese indigenous Laiwu pigs revealed candidate genes regulating fat deposition in muscle. BMC Genet. 2018, 19, 31. [Google Scholar] [CrossRef]
- Linah, A.A.; Muraly, P.; Kathy, R.; Mats, B.N.M.; James, A.; Jeffrey, B.; Ken, M.; Curry, T.E. Chemokine Ligand 20: A Signal for Leukocyte Recruitment During Human Ovulation? Endocrinology 2015, 156, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W.; et al. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. Gigascience 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Cui, L.; Perez-Enciso, M.; Traspov, A.; Rpma, C.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C.; et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Jakobsson, M.; Rosenberg, N.A. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 2008, 24, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Barbato, M.; Orozco-Terwengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Sved, J.A.; Feldman, M.W. Correlation and probability methods for one and two loci. Theor. Popul. Biol. 1973, 4, 129–132. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Long, Y.; Liao, X.; Ai, H.; Zhang, Z.; Yang, B.; Xiao, S.; Tang, J.; Xin, W.; Huang, L.; et al. Detection of genomic signatures for pig hairlessness using high-density SNP data. Front. Agric. Sci. Eng. 2014, 1, 307–313. [Google Scholar] [CrossRef]
- Liu, L.L.; Fang, C.; Liu, W.J. Identification on novel locus of dairy traits of Kazakh horse in Xinjiang. Gene 2018, 677, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2018, 47, D701–D710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Ramos, A.M.; Crooijmans, R.P.M.A.; Affara, N.A.; Amaral, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE 2009, 4, e6524. [Google Scholar] [CrossRef]
- Li, S.J.; Yang, S.L.; Zhao, S.H.; Fan, B.; Yu, M.; Wang, H.S.; Li, M.H.; Liu, B.; Xiong, T.A.; Li, K.; et al. Genetic diversity analyses of 10 indigenous Chinese pig populations based on 20 microsatellites. J. Anim. Sci. 2004, 82, 368–374. [Google Scholar] [CrossRef]
- Mujibi, F.D.; Okoth, E.; Cheruiyot, E.K.; Onzere, C.; Bishop, R.P.; Fèvre, E.M.; Thomas, L.; Masembe, C.; Plastow, G.; Rothschild, M.; et al. Genetic diversity, breed composition and admixture of Kenyan domestic pigs. PLoS ONE 2018, 13, e0190080. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Guo, J.F.; Zhang, Q.; Hu, H.M.; Lin, H.C.; Wang, C.; Zhang, Y.; Wu, Y. Genetic Diversity of Chinese Indigenous Pig Breeds in Shandong Province Using Microsatellite Markers. J. Anim. Sci. 2011, 24, 28–36. [Google Scholar] [CrossRef]
- Badke, Y.M.; Bates, R.O.; Ernst, C.W.; Schwab, C.; Steibel, J.P. Estimation of linkage disequilibrium in four US pig breeds. BMC Genom. 2012, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Grossi, D.A.; Jafarikia, M.; Brito, L.F.; Buzanskas, M.E.; Sargolzaei, M.; Schenkel, F.S. Genetic diversity, extent of linkage disequilibrium and persistence of gametic phase in Canadian pigs. BMC Genet. 2017, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zou, H.; Chen, L.; Long, X.; Lan, J.; Liu, W.; Ma, L.; Wang, C.; Xu, X.; Ren, L.; et al. Convergent and divergent genetic changes in the genome of Chinese and European pigs. Sci. Rep.-UK 2017, 7. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, J.B.; Xue, Y.; Peng, Y.L.; Chen, G.; Fang, M.Y. Differential expression of CYB5A in Chinese and European pig breeds due to genetic variations in the promoter region. Anim. Genet. 2015, 46, 16–22. [Google Scholar] [CrossRef]
- Fang, M.; Larson, G.; Ribeiro, H.S.; Li, N.; Andersson, L. Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genet. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Kijas, J.M.H.; Wales, R.; Tornsten, A.; Chardon, P.; Moller, M.; Andersson, L. Melanocortin receptor 1 (MC1R) mutations and coat color in pigs. Genetics 1998, 150, 1177–1185. [Google Scholar]
- Kobayashi, Y.; Hamamoto, A.; Takahashi, A.; Saito, Y. Dimerization of melanocortin receptor 1 (MC1R) and MC5R creates a ligand-dependent signal modulation: Potential participation in physiological color change in the flounder. Gen. Comp. Endocrinol. 2016, 230, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-E.; Hwangbo, Y.; Kim, H.-Y.; Lee, W.-H.; Cheong, H.-T.; Yang, B.-K.; Park, C.-K. Effect of Alpha-Linolenic Acid on Oocyte Maturation and Embryo Development in Pigs. Dev. Reprod. 2017, 21, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, B.N.; Fraser, D.; Kramer, D.L. Within-litter birth weight variation in the domestic pig and its relation to pre-weaning survival, weight gain, and variation in weaning weights. Livest. Prod. Sci. 2002, 76, 181–191. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed 1 | Origin | No. | Sex Composition, Male/Female/Ambiguous | NSNP 2 | PN 3 | HO 4 | HE 5 | AR 6 | Ne 7 |
|---|---|---|---|---|---|---|---|---|---|
| DRC | - | 79 | 8/12/59 | 8010 | 0.998 | 0.305 | 0.334 | 1.940 | 67.920 |
| LW | - | 76 | 8/12/56 | 10,135 | 0.998 | 0.363 | 0.400 | 1.998 | 76.960 |
| LR | - | 130 | 10/10/110 | 10,483 | 0.998 | 0.371 | 0.407 | 1.998 | 81.230 |
| PI | - | 58 | 0/0/58 | 9481 | 0.993 | 0.373 | 0.386 | 1.995 | 48.760 |
| WBE | - | 153 | 20/0/133 | 7893 | 1.000 | 0.251 | 0.319 | 1.913 | 150.530 |
| WBA | - | 63 | 18/8/37 | 9876 | 1.000 | 0.338 | 0.390 | 1.996 | 73.080 |
| BMX | Guangxi | 26 | 8/18/0 | 8426 | 1.000 | 0.365 | 0.351 | 1.971 | 26.450 |
| DB | Guangxi | 15 | 1/14/0 | 9329 | 0.984 | 0.371 | 0.368 | 1.966 | 16.260 |
| DHB | Guangxi | 32 | 3/29/0 | 7047 | 1.000 | 0.343 | 0.311 | 1.942 | 15.700 |
| GZH | Guangxi | 29 | 5/24/0 | 8348 | 1.000 | 0.366 | 0.348 | 1.962 | 32.020 |
| LC | Guangxi | 28 | 3/25/0 | 7237 | 0.984 | 0.328 | 0.315 | 1.898 | 34.930 |
| LL | Guangdong | 25 | 3/22/0 | 11,008 | 0.984 | 0.430 | 0.420 | 1.999 | 27.890 |
| LT | Guangdong | 28 | 6/22/0 | 7406 | 1.000 | 0.351 | 0.320 | 1.906 | 17.400 |
| MH | Guangdong | 24 | 6/18/0 | 7847 | 1.000 | 0.360 | 0.327 | 1.895 | 11.020 |
| XE | Guangdong | 34 | 0/34/0 | 8299 | 0.984 | 0.363 | 0.350 | 1.973 | 45.410 |
| YDH | Guangdong | 25 | 6/19/0 | 10,400 | 1.000 | 0.432 | 0.399 | 1.990 | 15.910 |
| Chr | ID | SNP | Position, bp | FST | Genes | QTL 2 (Counts) |
|---|---|---|---|---|---|---|
| 1 | rs80947145 | ASGA0004899 | 149,872,523 | 0.852 | C1H18orf63, CYB5A, TIMM21, FBXO15 | Drip loss (15) |
| 8 | rs81261671 | MARC0076384 | 54,466,796 | 0.845 | -- | Mean corpuscular volume (3) |
| 8 | rs81339817 | ALGA0111390 | 55,931,429 | 0.838 | SRP72, ARL9, THEGL, HOPX, SPINK2, REST | Mean corpuscular volume (4) |
| 5 | rs81383780 | ALGA0031742 | 36,506,086 | 0.834 | TRHDE | Meat color b* (4) |
| 5 | rs80983312 | MARC0046863 | 44,931,786 | 0.831 | -- | Average backfat thickness (4) |
| 7 | rs80995372 | ALGA0045445 | 125,434,386 | 0.825 | -- | Shoulder subcutaneous fat thickness (3) |
| Term | Count | Genes | p-Value |
|---|---|---|---|
| ssc00592:alpha-Linolenic acid metabolism | 6 | PLA2G12A, PLA2G12B, PLA2G2C, PLA2G2D, PLA2G5, PLA2G2F | 0.001 |
| ssc04975:Fat digestion and absorption | 7 | PLA2G12A, PLA2G12B, FABP1, PLA2G2C, PLA2G2D, PLA2G5, PLA2G2F | 0.002 |
| GO:0050482~arachidonic acid secretion | 5 | PLA2G12A, PLA2G12B, PLA2G2C, PLA2G2D, PLA2G2F | 0.002 |
| GO:0007338~single fertilization | 7 | PSP-II, RNASE10, PSP-I, AWN, AQN-1, SMAD4, SPMI | 0.003 |
| GO:0090501~RNA phosphodiester bond hydrolysis | 3 | ANG, RNASE4, RNASE6 | 0.005 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diao, S.; Huang, S.; Xu, Z.; Ye, S.; Yuan, X.; Chen, Z.; Zhang, H.; Zhang, Z.; Li, J. Genetic Diversity of Indigenous Pigs from South China Area Revealed by SNP Array. Animals 2019, 9, 361. https://doi.org/10.3390/ani9060361
Diao S, Huang S, Xu Z, Ye S, Yuan X, Chen Z, Zhang H, Zhang Z, Li J. Genetic Diversity of Indigenous Pigs from South China Area Revealed by SNP Array. Animals. 2019; 9(6):361. https://doi.org/10.3390/ani9060361
Chicago/Turabian StyleDiao, Shuqi, Shuwen Huang, Zhiting Xu, Shaopan Ye, Xiaolong Yuan, Zanmou Chen, Hao Zhang, Zhe Zhang, and Jiaqi Li. 2019. "Genetic Diversity of Indigenous Pigs from South China Area Revealed by SNP Array" Animals 9, no. 6: 361. https://doi.org/10.3390/ani9060361