RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Adipose Tissue Sample
2.2. RNA Extraction and RNA Sequencing
2.3. Sequencing Data Analysis
2.4. GO Enrichment and KEGG Pathway Analysis
2.5. Real-Time Quantitative PCR Analysis
2.6. Statistical Analysis
3. Results
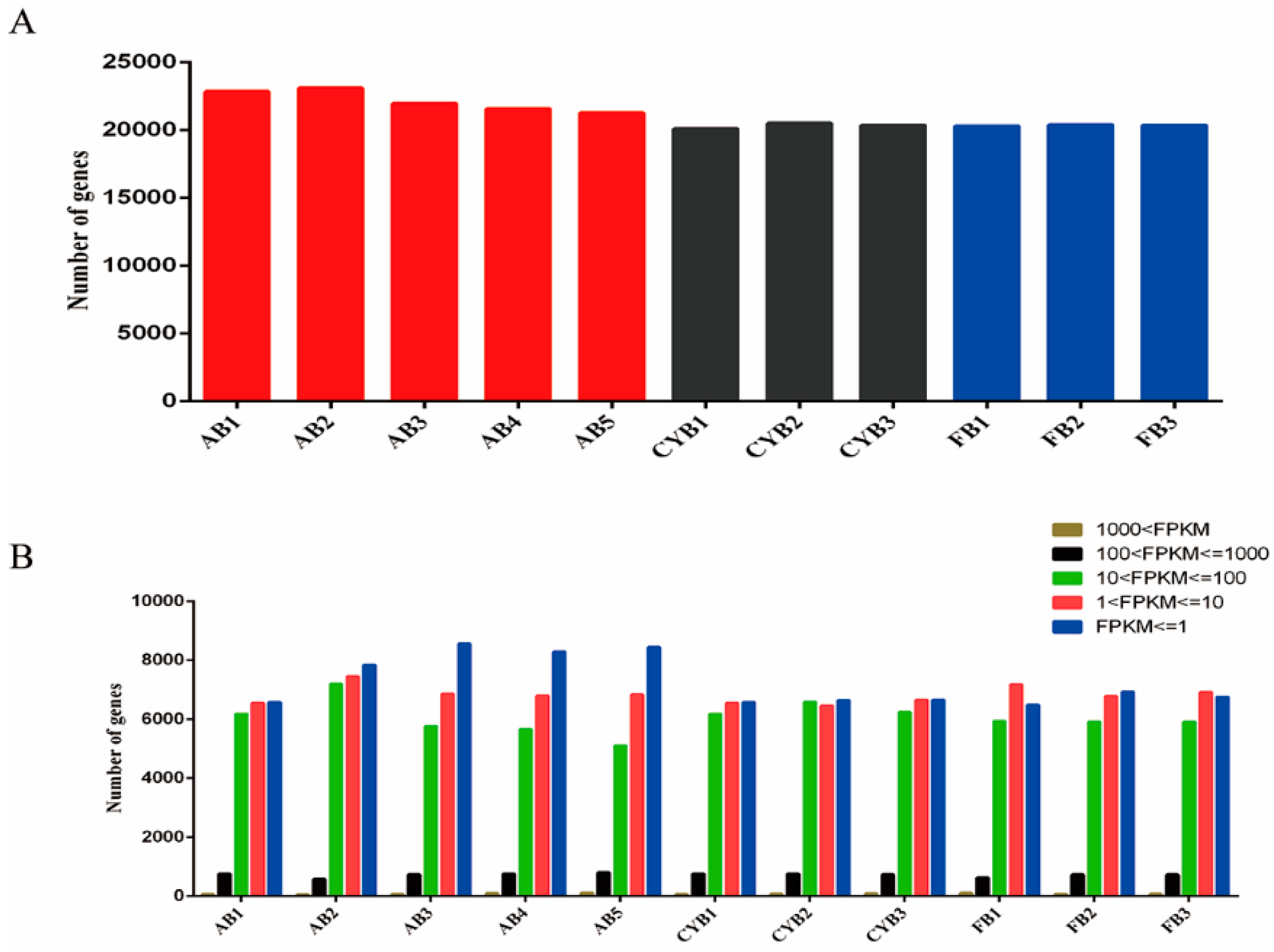
3.1. Characterization of Bovine Adipose Tissue Transcriptome Sequencing Data
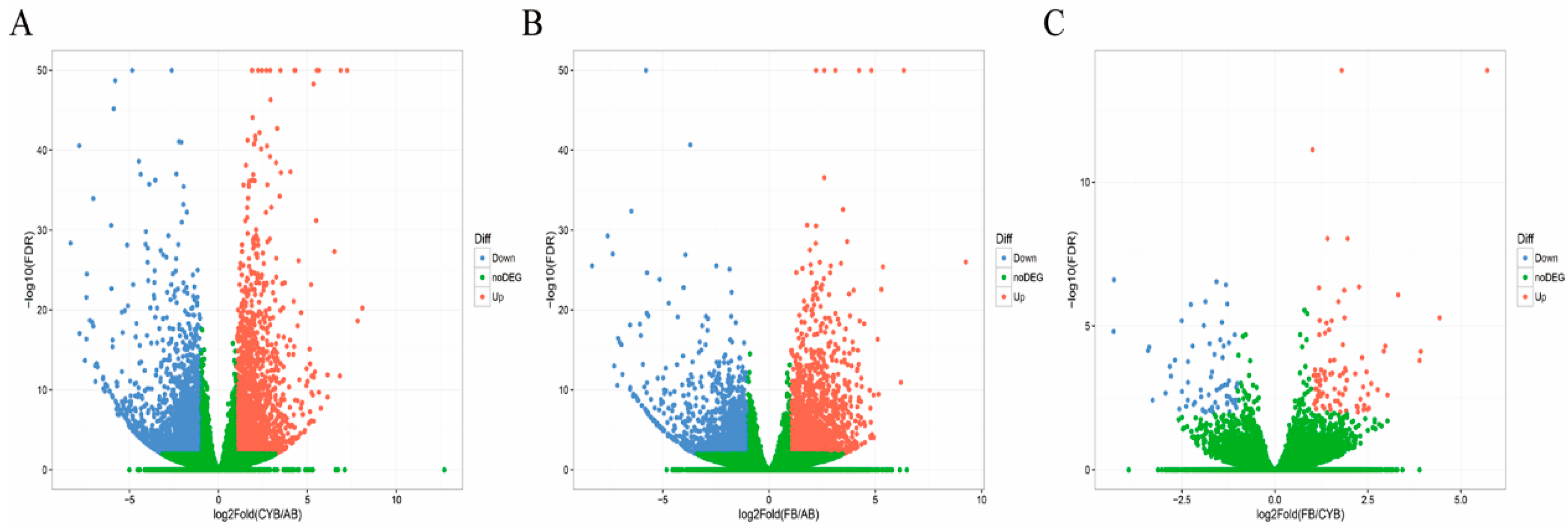
3.2. Identification of Differentially Expressed Genes (DEGs)
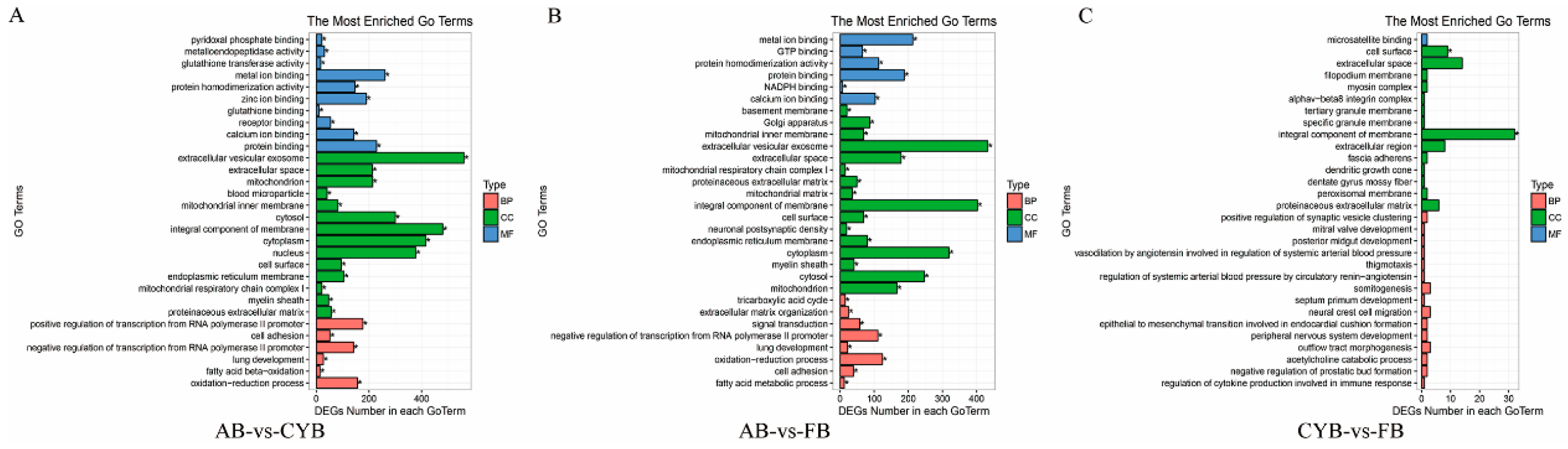
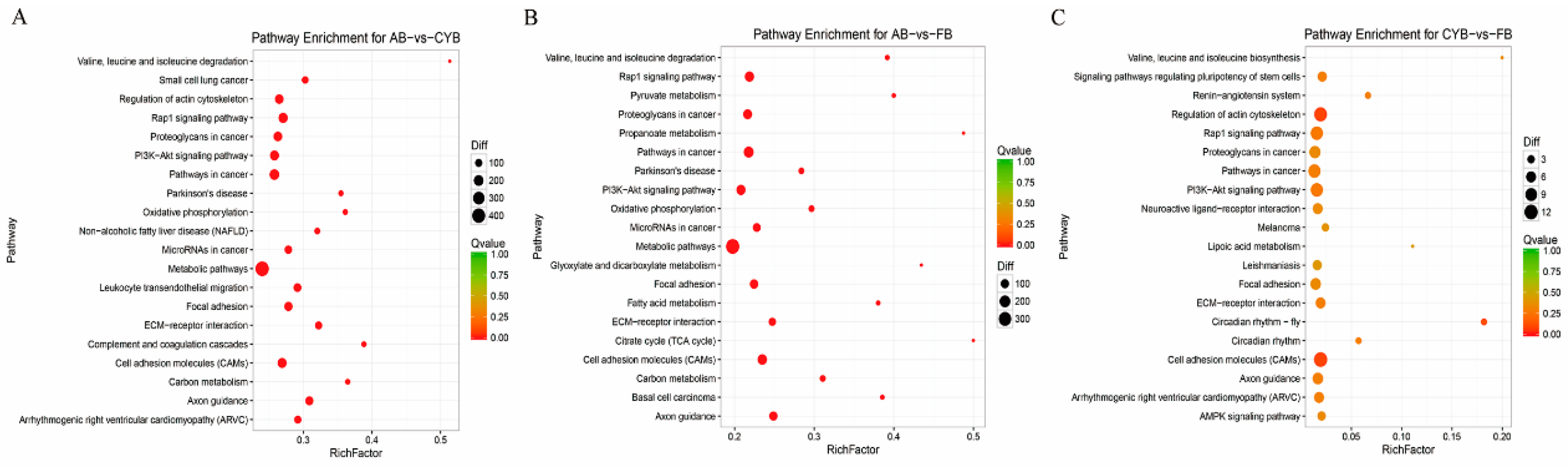
3.3. GO Enrichment and KEGG Pathway Analysis of Related DEGs
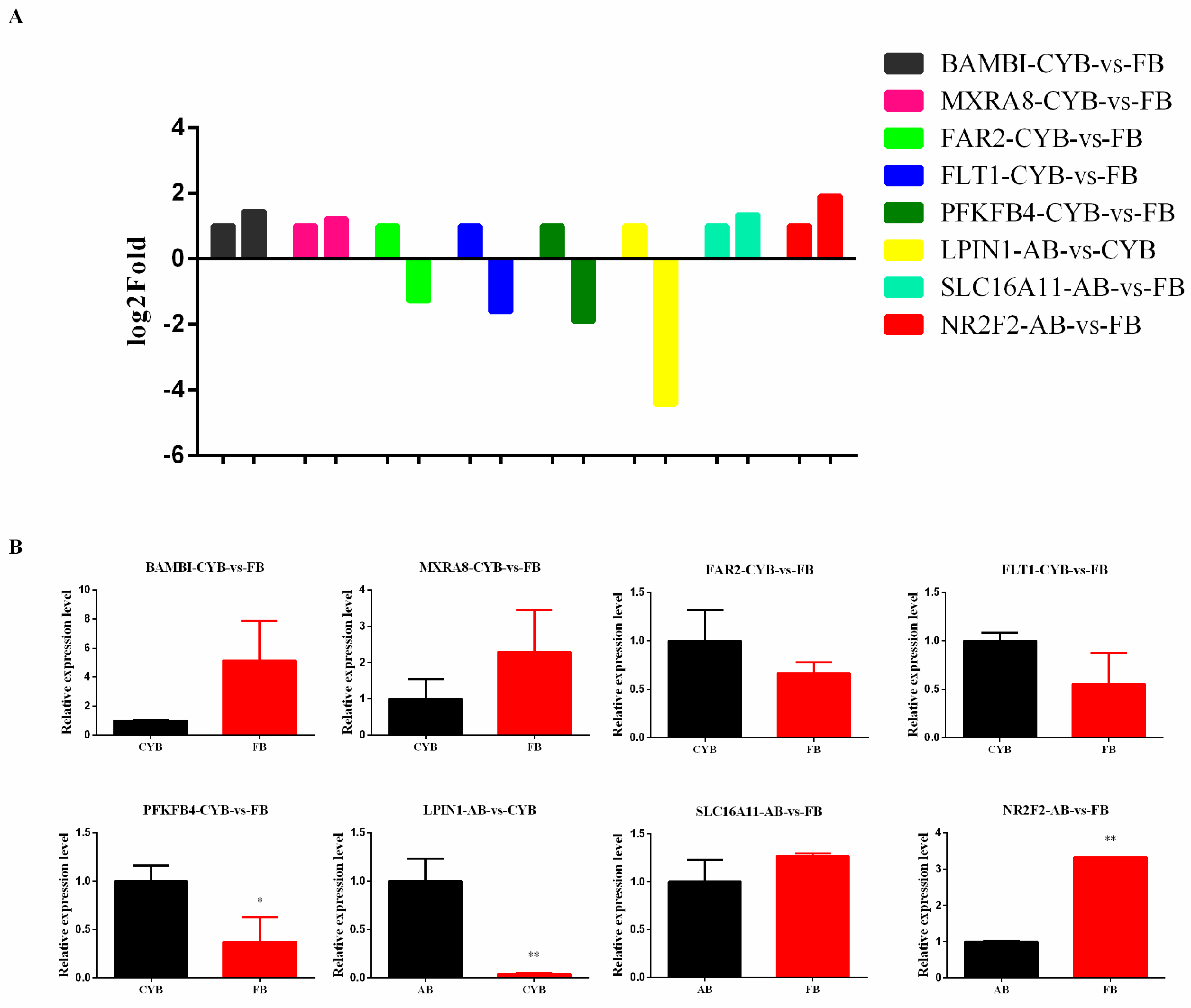
3.4. DEGs Were Validated Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feng, Y.; Liu, Y.; Xie, P.; Lang, Y.; Haipeng, L.; Baozhong, S. Meat Quality Characteristics and Safety Analysis of Qaidamford Cattle. Meat Res. 2015, 29, 6–10. [Google Scholar]
- Zheng, Y.; Wang, S.; Yan, P. The meat quality, muscle fiber characteristics and fatty acid profile in Jinjiang and F1 SimmentalxJinjiang yellow cattle. Asian Australas. J. Anim. Sci. 2018, 31, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.D.; Enser, M.; Fisher, A.V.; Nute, G.R.; Sheard, P.R.; Richardson, R.I.; Hughes, S.I.; Whittington, F.M. Fat deposition, fatty acid composition and meat quality: A review. Meat Sci. 2008, 78, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Finelli, C.; Sommella, L.; Gioia, S.; La Sala, N.; Tarantino, G. Should visceral fat be reduced to increase longevity? Ageing Res. Rev. 2013, 12, 996–1004. [Google Scholar] [CrossRef]
- Stewart, W.C.; Pearcy, L.A.; Floyd, Z.E.; Stephens, J.M. STAT5A expression in Swiss 3T3 cells promotes adipogenesis in vivo in an athymic mice model system. Obesity 2011, 19, 1731–1734. [Google Scholar] [CrossRef] [Green Version]
- White, U.A.; Stephens, J.M. Transcriptional factors that promote formation of white adipose tissue. Mol. Cell. Endocrinol. 2010, 318, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Mota de Sa, P.; Richard, A.J.; Hang, H.; Stephens, J.M. Transcriptional Regulation of Adipogenesis. Compr. Physiol. 2017, 7, 635–674. [Google Scholar]
- Huang, W.; Khatib, H. Comparison of transcriptomic landscapes of bovine embryos using RNA-Seq. BMC Genom. 2010, 11, 711. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Guo, Y.; Du, W.; Zhang, X.; Li, A.; Miao, X. Global transcriptome analysis identifies differentially expressed genes related to lipid metabolism in Wagyu and Holstein cattle. Sci. Rep. 2017, 7, 5278. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.; Li, S.; Abbas, S.H.; Tian, W.; Wang, H.; Li, Y.; Gui, L.; Zhang, Y.; Wu, X.; Zan, L. Performance Measurement and Comparative Transcriptome Analysis Revealed the Efforts on Hybrid Improvement of Qinchuan Cattle. Anim. Biotechnol. 2018, 30, 13–20. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: Hierarchical Indexing for Spliced Alignment of Transcripts. Bioinformatics 2014. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamimuzzaman, M.; Le Tourneau, J.J.; Unni, D.R.; Diesh, C.M.; A Triant, D.; Walsh, A.T.; Tayal, A.; Conant, G.C.; E Hagen, D.; Elsik, C.G. Bovine Genome Database: New annotation tools for a new reference genome. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Marguerat, S.; Bähler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Ayers, K.L.; Lambeth, L.S.; Davidson, N.M.; Sinclair, A.H.; Oshlack, A.; Smith, C.A. Identification of candidate gonadal sex differentiation genes in the chicken embryo using RNA-seq. BMC Genom. 2015, 16, 704. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Guo-Qin, L.I.; Tian, Y.; Shen, J.D.; Tao, Z.R.; Xu, J.; Zeng, T.; Lu, L.Z. Transcriptome analysis of abdominal fats from Peking ducks by RNA-seq. Acta Agric. Zhejiangensis 2016, 28, 743–747. [Google Scholar]
- Ropka-Molik, K.; Zukowski, K.; Eckert, R.; Gurgul, A.; Piórkowska, K.; Oczkowicz, M. Comprehensive analysis of the whole transcriptomes from two different pig breeds using RNA-Seq method. Anim. Genet. 2015, 45, 674–684. [Google Scholar] [CrossRef]
- Sheng, X.; Ni, H.; Liu, Y.; Li, J.; Zhang, L.; Guo, Y. RNA-seq analysis of bovine intramuscular, subcutaneous and perirenal adipose tissues. Mol. Biol. Rep. 2014, 41, 1631–1637. [Google Scholar]
- Kuryłowicz, A.; Wicik, Z.; Owczarz, M.; Jonas, M.I.; Kotlarek, M.; Świerniak, M.; Lisik, W.; Jonas, M.; Noszczyk, B.; Puzianowska-Kuźnicka, M. NGS Reveals Molecular Pathways Affected by Obesity and Weight Loss-Related Changes in miRNA Levels in Adipose Tissue. Int. J. Mol. Sci. 2018, 19, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Cui, X.; Shi, C.; Chen, L.; Yang, L.; Pang, L.; Zhang, J.; Guo, X.; Wang, J.; Ji, C. Differential lncRNA expression profiles in brown and white adipose tissues. Mol. Genet. Genom. MGG 2015, 290, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, F.; Zhang, Y.; Dai, L.; Jiang, H.; Liu, H.; Zhang, S.; Chen, C.; Zhang, J. Silencing of ADIPOQ Efficiently Suppresses Preadipocyte Differentiation in Porcine. Cell. Physiol. Biochem. 2013, 31, 452–461. [Google Scholar] [CrossRef]
- Lee, H.-J.; Jang, M.; Kim, H.; Kwak, W.; Park, W.; Hwang, J.Y.; Lee, C.-K.; Jang, G.W.; Na Park, M.; Kim, H.-C.; et al. Comparative Transcriptome Analysis of Adipose Tissues Reveals that ECM-Receptor Interaction Is Involved in the Depot-Specific Adipogenesis in Cattle. PLoS ONE 2013, 8, e66267. [Google Scholar] [CrossRef]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Jin, H.; Cao, Y.; Zhang, L.; Zhao, Y.; Jin, X.; Yu, Y. RNA-Seq Analysis Reveals a Positive Role of HTR2A in Adipogenesis in Yan Yellow Cattle. Int. J. Mol. Sci. 2018, 19, 1760. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.P.; Ha, J.M.; Yun, S.J.; Kim, E.K.; Chung, S.W.; Hong, K.W.; Kim, C.D.; Bae, S.S. Transcriptional activation of peroxisome proliferator-activated receptor-gamma requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2010, 399, 55–59. [Google Scholar] [CrossRef]
- Luo, X.; Hutley, L.J.; Webster, J.A.; Kim, Y.H.; Liu, D.F.; Newell, F.S.; Widberg, C.H.; Bachmann, A.; Turner, N.; Schmitz-Peiffer, C.; et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI) as a potent negative regulator of adipogenesis and modulator of autocrine/paracrine adipogenic factors. Diabetes 2012, 61, 124–136. [Google Scholar] [CrossRef] [Green Version]
- The SIGMA Type 2 Diabetes Consortium. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 2014, 506, 97–101. [Google Scholar] [CrossRef]
- Rusu, V.; Hoch, E.; Tenen, D.E.; Gymrek, M.; Hartigan, C.R.; DeRan, M.; Von Grotthuss, M.; Fontanillas, P.; Spooner, A.; Guzman, G.; et al. Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 2017, 170, 199–212.e20. [Google Scholar] [CrossRef]
- Heckman, M. The Circadian Clock and Nr2f2 (COUP-TF) Regulation of Adipogenesis; University of Rochester: Rochester, NY, USA, 2008. [Google Scholar]
- Clark, D.L.; Boler, D.D.; Kutzler, L.W.; Jones, K.A.; McKeith, F.K.; Killefer, J.; Carr, T.R.; Dilger, A.C. Muscle gene expression associated with increased marbling in beef cattle. Anim. Biotechnol. 2011, 22, 51–63. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Xu, X.W.; Zhao, S.H.; Fan, B.; Yu, M.; Zhu, M.J.; Li, C.C.; Peng, Z.Z.; Liu, B. Investigation of Lpin1 as a candidate gene for fat deposition in pigs. Mol. Biol. Rep. 2009, 36, 1175–1180. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Huang, Y.; Yang, Z.; Ma, Y.; Chaogetu, B.; Zhuoma, Z.; Chen, H. RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle. Animals 2019, 9, 1077. https://doi.org/10.3390/ani9121077
Song C, Huang Y, Yang Z, Ma Y, Chaogetu B, Zhuoma Z, Chen H. RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle. Animals. 2019; 9(12):1077. https://doi.org/10.3390/ani9121077
Chicago/Turabian StyleSong, Chengchuang, Yongzhen Huang, Zhaoxin Yang, Yulin Ma, Buren Chaogetu, Zhaxi Zhuoma, and Hong Chen. 2019. "RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle" Animals 9, no. 12: 1077. https://doi.org/10.3390/ani9121077