
Comparison of Intestinal Bacteria of Procambarus clarkii Farmed in Various Rice Paddy Regions
 , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Pretreatment
2.2. DNA Extraction, PCR Amplification, and Sequencing DNA
2.3. Statistical Analysis
3. Results
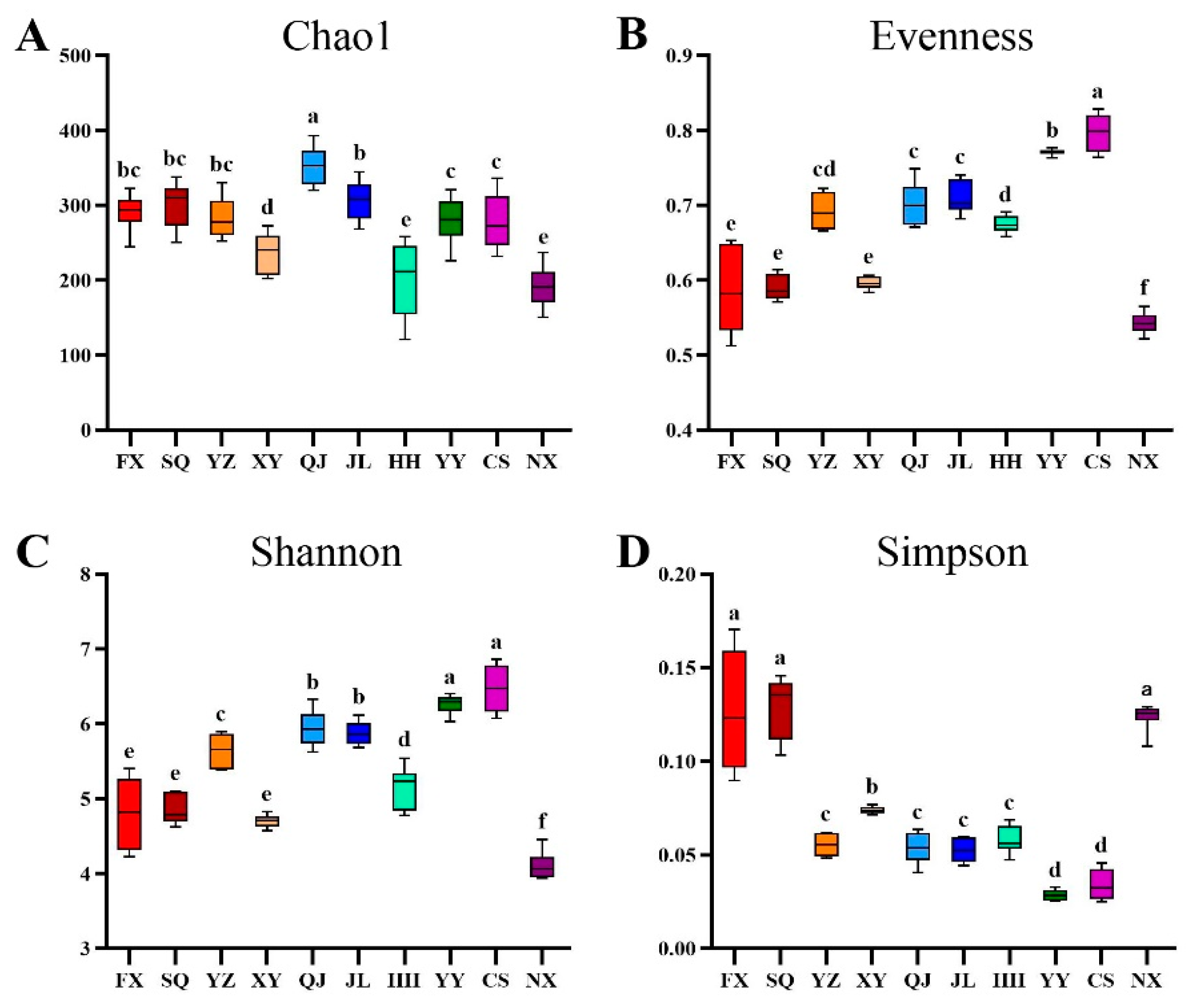
3.1. Intestinal Microbiota Alpha-Diversity Analysis
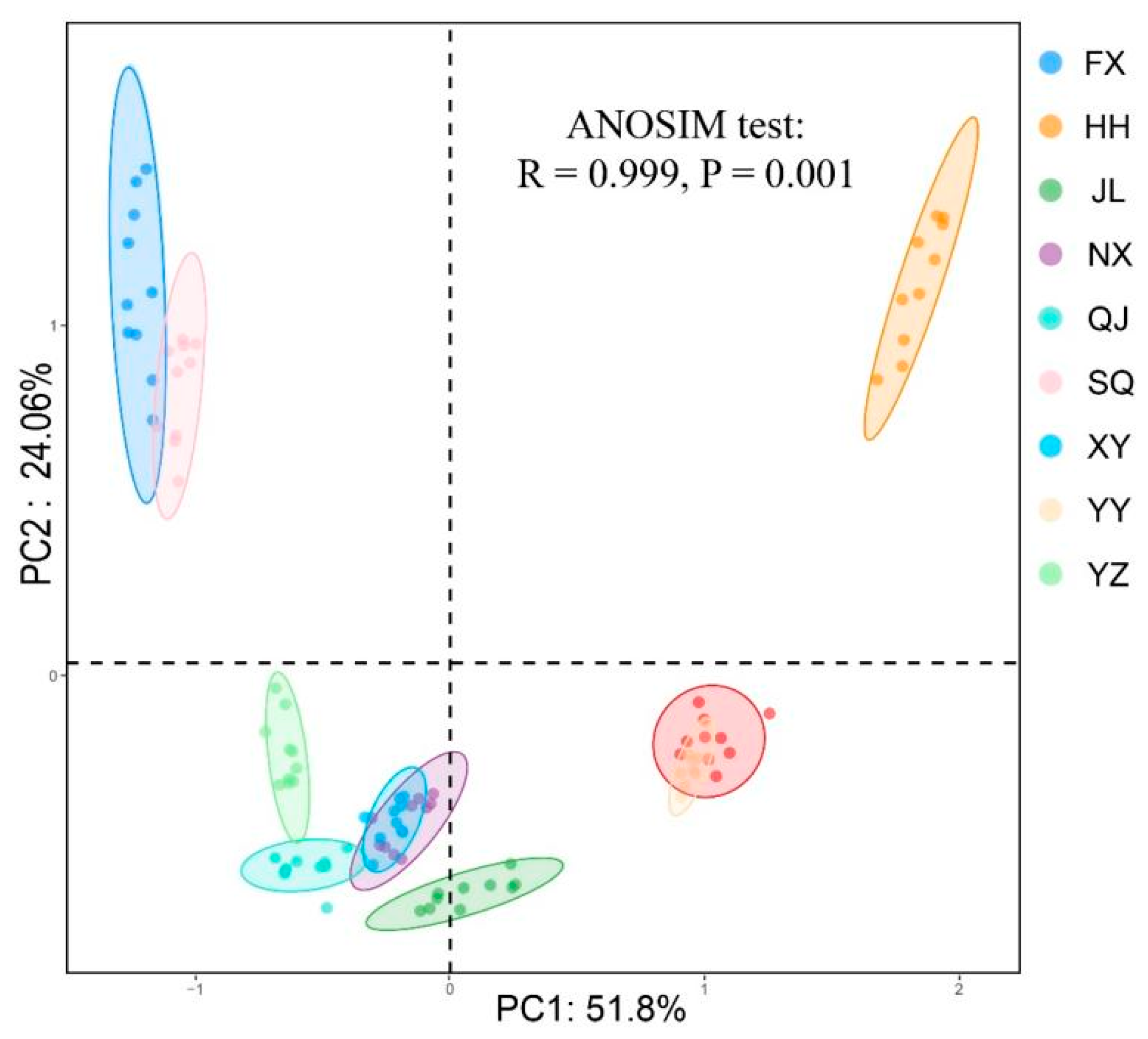
3.2. Intestinal Microbiota Beta-Diversity Analysis
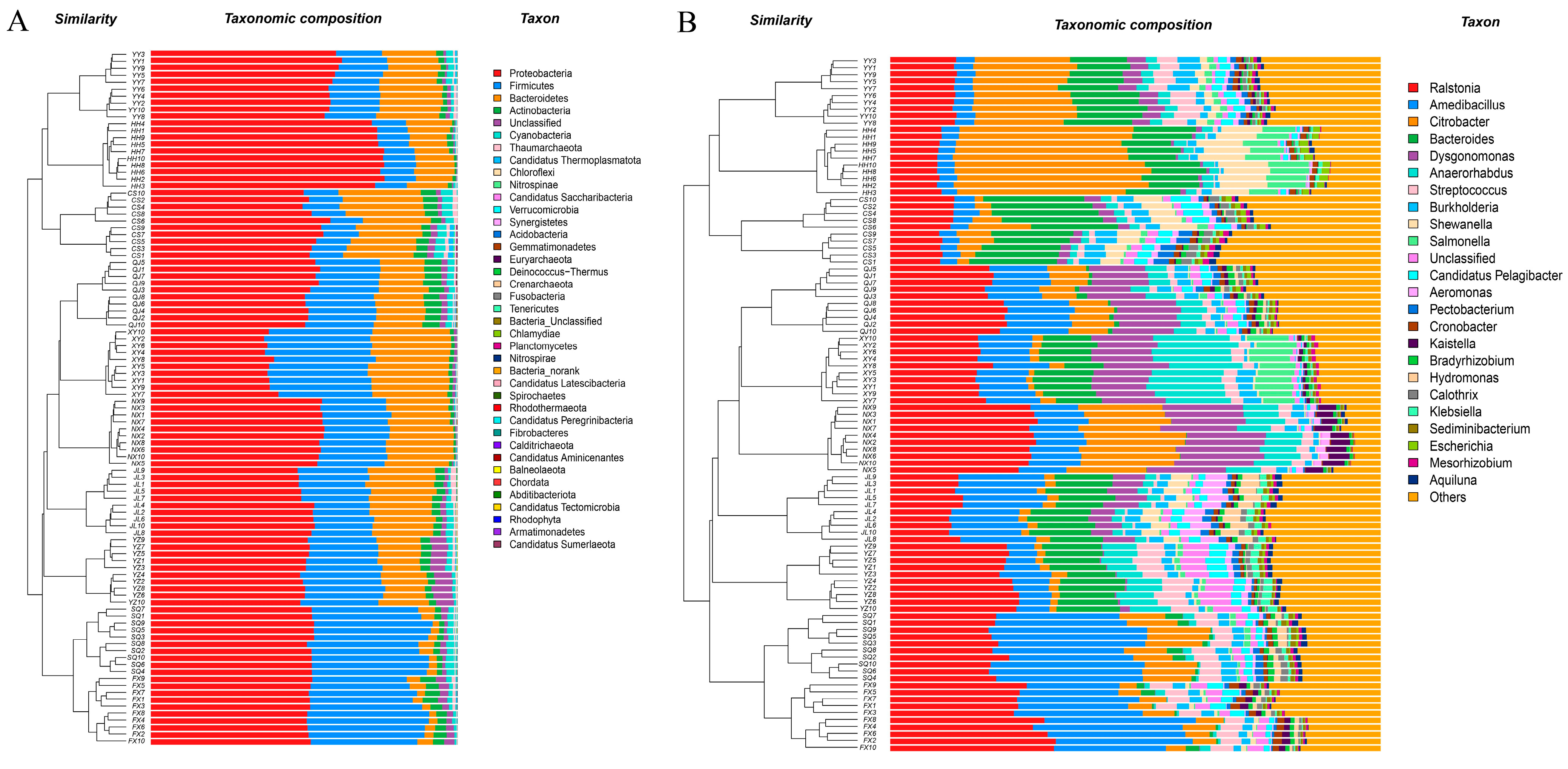
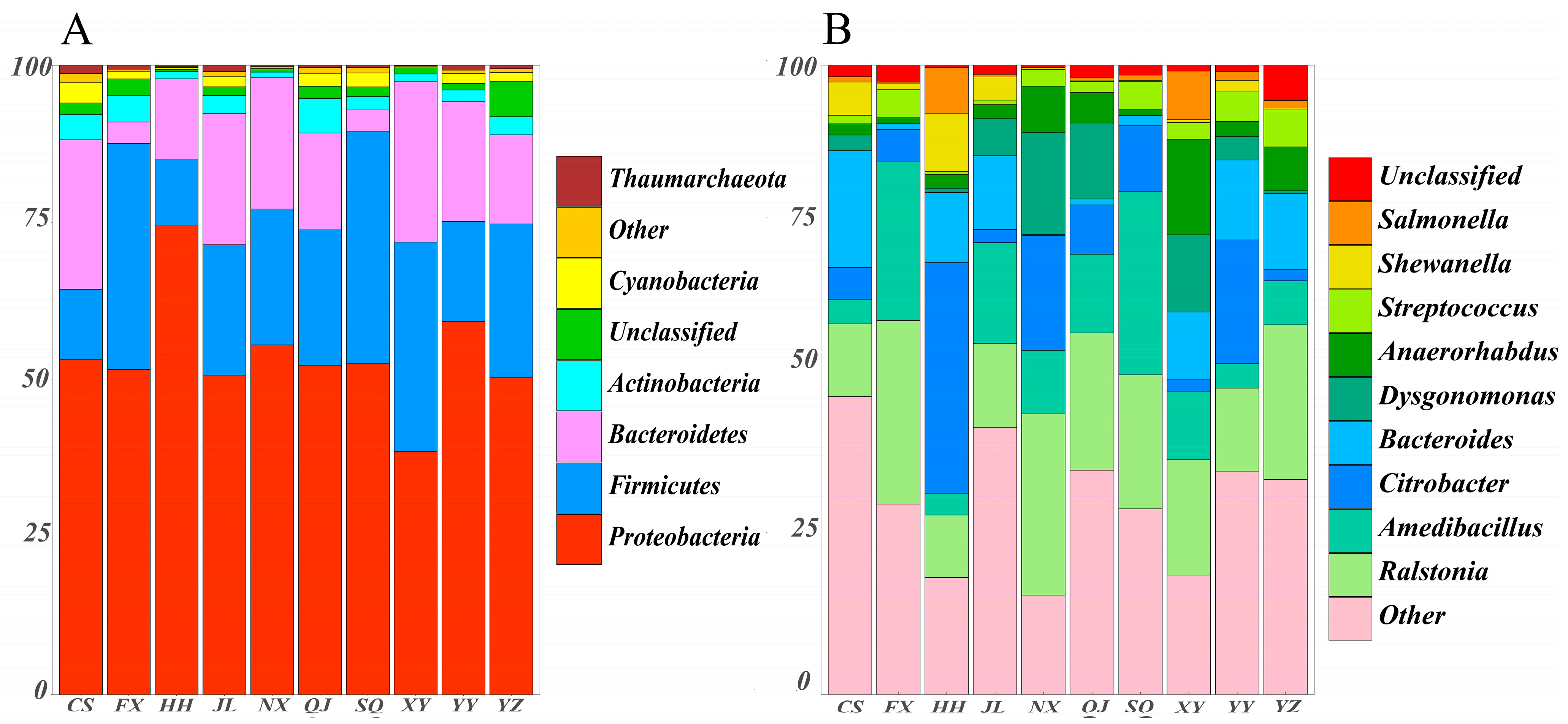
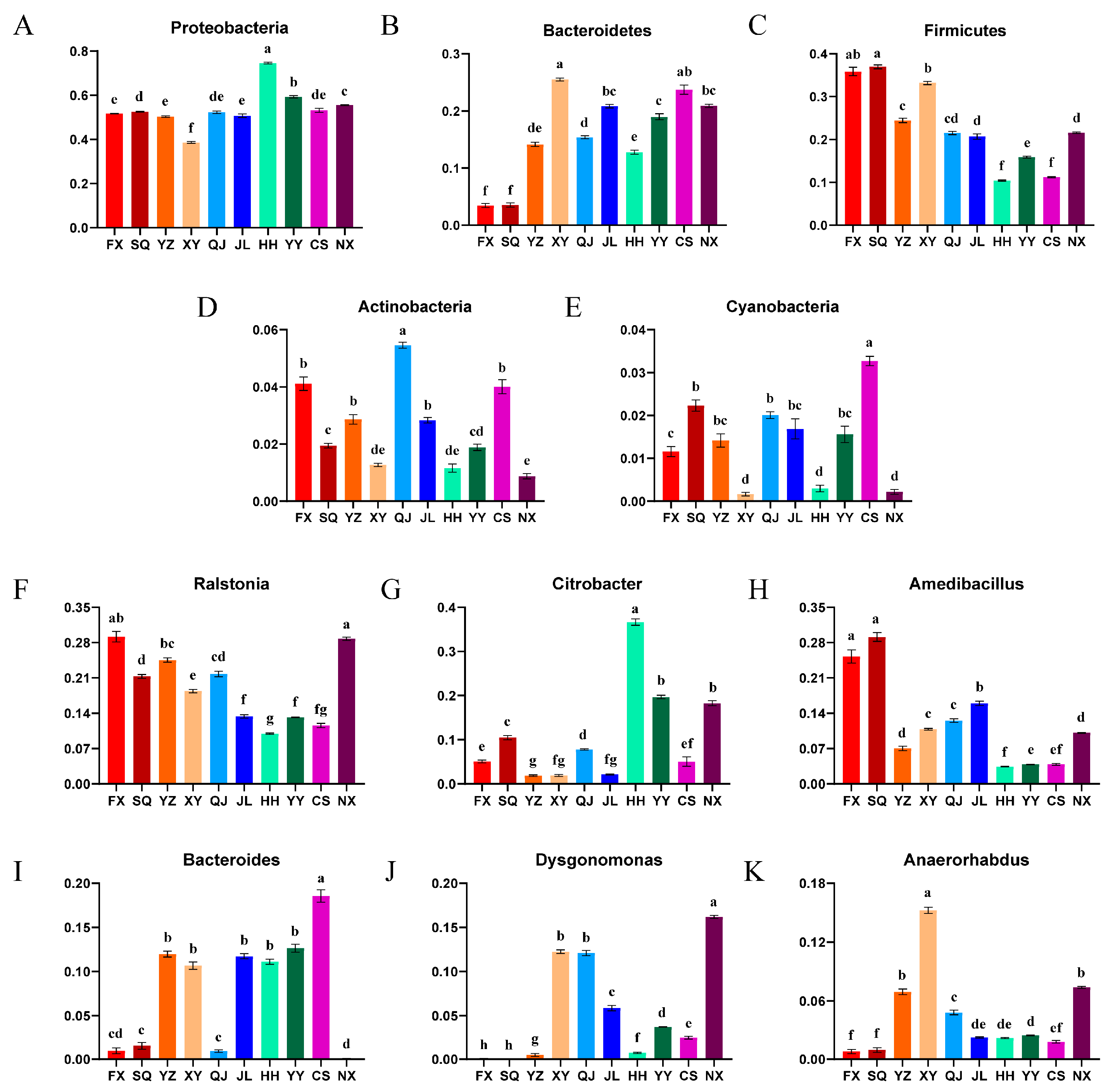
3.3. Microbial Community Composition
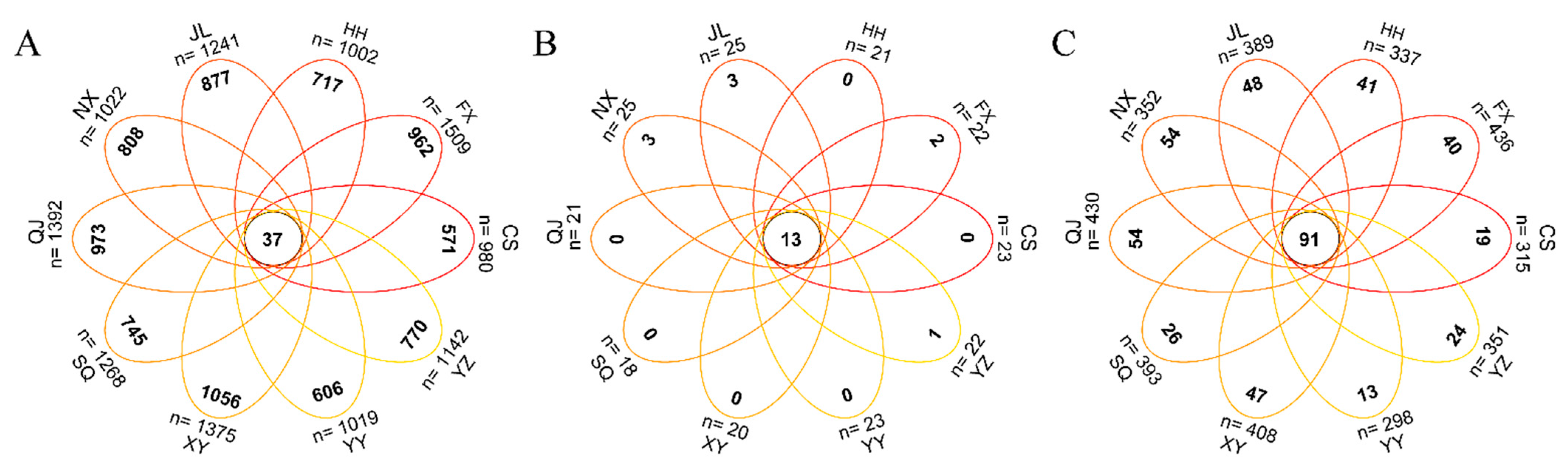
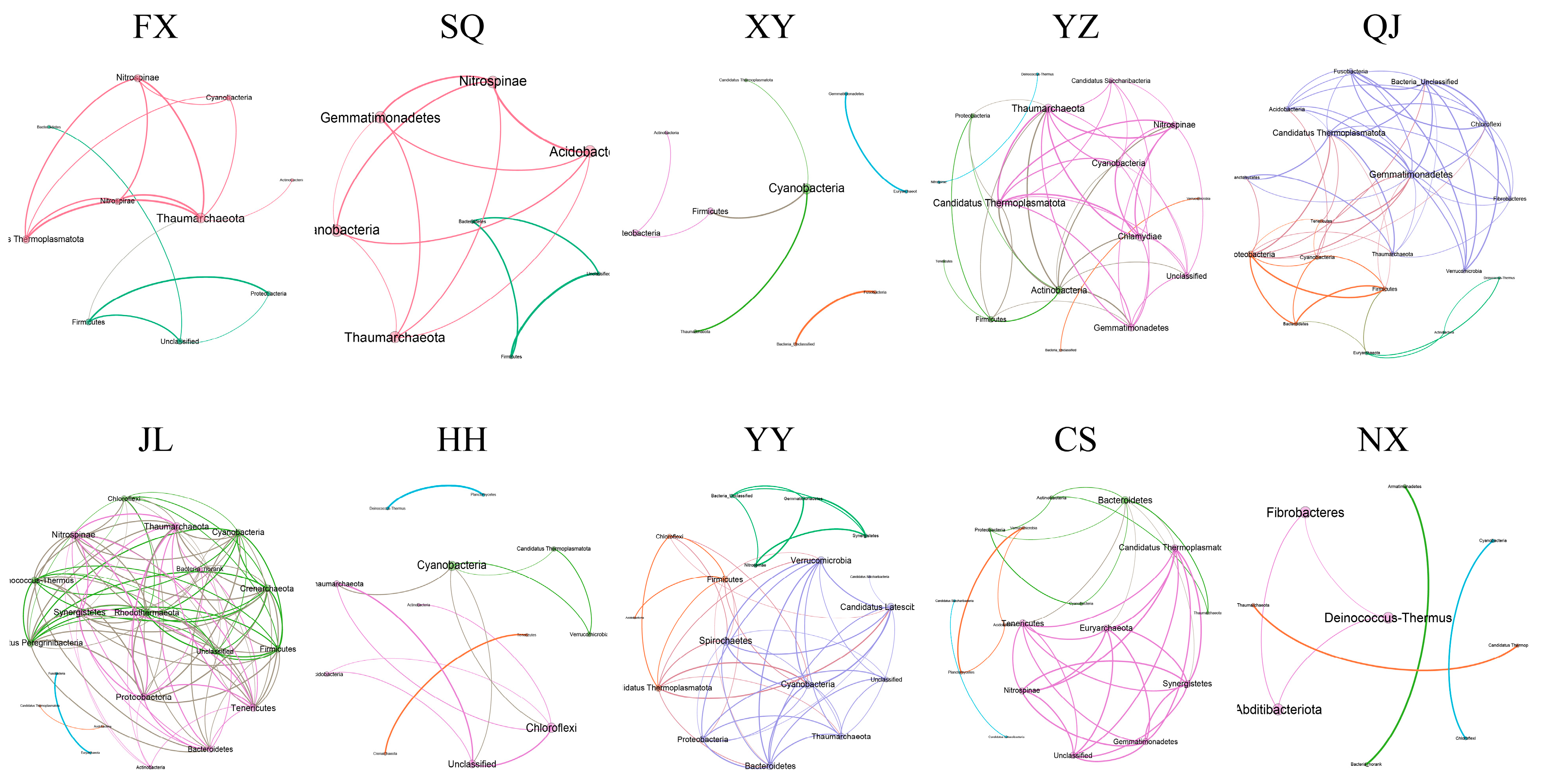
3.4. Co-Occurrence Networks of P. clarkii Intestinal Bacterial Communities in Different Areas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bi, K.; Gu, W.; Wang, W. Sensitive and rapid detection of freshwater crustacean Spiroplasmas by ISRs-sequence-targeted species-specific primers. Eur. Food Res. Technol. 2008, 227, 1733–1737. [Google Scholar] [CrossRef]
- Li, Y.; Guo, X.; Cao, X.; Deng, W.; Luo, W.; Wang, W. Population genetic structure and post-establishment dispersal patterns of the red swamp crayfish Procambarus clarkii in China. PLoS ONE 2012, 7, e40652. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hao, X.; Dang, Z.; Yang, L. China crayfish industry development report. China Fish. 2023, 7, 26–31. [Google Scholar]
- Zheng, D.; Peng, X.; Zhang, X.; Zhang, J.; Zhou, Y.; Peng, L.; Xia, Z. Comparative Analysis on Muscle Quality of Procambarus clarkii Under Different Aquaculture Models in Hubei Province. Sci. Technol. Food Ind. 2023, 44, 91–97. [Google Scholar] [CrossRef]
- Shui, Y.; Guan, Z.B.; Liu, G.F.; Fan, L.M. Gut microbiota of red swamp crayfish Procambarus clarkii in integrated crayfish-rice cultivation model. AMB Express 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cao, C. Crayfish–rice integrated system of production: An agriculture success story in China. A review. Agron. Sustain. Dev. 2021, 41, 68. [Google Scholar] [CrossRef]
- Nie, Z.; Xu, X.; Shao, N.; He, J.; Li, P.; Xu, P.; Hu, J.; Qin, W.; Wang, B.; Xu, G. Integrative analysis of microbiome and metabolome reveals the linkage between gut microbiota and carp growth. Environ. Res. 2023, 220, 115133. [Google Scholar] [CrossRef] [PubMed]
- Si, G.; Peng, C.; Xu, X.; Xu, D.; Yuan, J.; Li, J. Effect of integrated rice-crayfish farming system on soil physico-chemical properties in waterlogged paddy soils. Chin. J. Eco-Agric. 2017, 25, 61–68. [Google Scholar] [CrossRef]
- Yu, X.; Hao, X.; Dang, Z.; Yang, L. Report on the Development of China’s Rice-Fishery Comprehensive Breeding Industry (2023). China Fish. 2023, 8, 19–26. [Google Scholar]
- Jia, L.; Wang, G.; Xia, Y.; Zhang, K.; Gao, S.; Li, Y.; Gao, Y. Comparison of physiological metabolism, muscle quality and nutritional value of Procambarus clarkii cultivated in paddy fields in different areas. J. Gansu Agric. Univ. 2022, 57, 188–197. [Google Scholar]
- Sun, Y.; Han, W.; Liu, J.; Huang, X.; Zhou, W.; Zhang, J.; Cheng, Y. Bacterial community compositions of crab intestine, surrounding water, and sediment in two different feeding modes of Eriocheir sinensis. Aquac. Rep. 2020, 16, 100236. [Google Scholar] [CrossRef]
- Huang, Z.; Li, X.; Wang, L.; Shao, Z. Changes in the intestinal bacterial community during the growth of white shrimp, Litopenaeus vannamei. Aquac. Res. 2016, 47, 1737–1746. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, M.; Liu, J.; Qiao, Y.; Wang, L.; Li, Z.; Zhang, X.-H.; Yu, M. Bacterial Community Associated with Healthy and Diseased Pacific White Shrimp (Litopenaeus vannamei) Larvae and Rearing Water across Different Growth Stages. Front. Microbiol. 2017, 8, 1362. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wang, Z.; Chen, M.; Qu, Y.; Li, J.; Zhou, A.; Xie, S.; Zeng, F.; Zou, J. Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 2019, 657, 1194–1204. [Google Scholar] [CrossRef]
- Tao, H.; Du, B.; Wang, H.; Dong, H.; Yu, D.; Ren, L.; Sima, Y.; Xu, S. Intestinal microbiome affects the distinctive flavor of Chinese mitten crabs in commercial farms. Aquaculture 2018, 483, 38–45. [Google Scholar] [CrossRef]
- Liu, Q.; Long, Y.; Li, B.; Zhao, L.; Luo, J.; Xu, L.; Luo, W.; Du, Z.; Zhou, J.; Yang, S. Rice-shrimp culture: A better intestinal microbiota, immune enzymatic activities, and muscle relish of crayfish (Procambarus clarkii) in Sichuan Province. Appl. Microbiol. Biotechnol. 2020, 104, 9413–9420. [Google Scholar] [CrossRef]
- Yuan, P.; Wang, J.; Li, C.; Xiao, Q.; Liu, Q.; Sun, Z.; Wang, J.; Cao, C. Soil quality indicators of integrated rice-crayfish farming in the Jianghan Plain, China using a minimum data set. Soil Tillage Res. 2020, 204, 104732. [Google Scholar] [CrossRef]
- Si, G.; Peng, C.; Yuan, J.; Xu, X.; Zhao, S.; Xu, D.; Wu, J. Changes in soil microbial community composition and organic carbon fractions in an integrated rice-crayfish farming system in subtropical China. Sci. Rep. 2017, 7, 2856. [Google Scholar] [CrossRef]
- Zhou, H.; Ge, T.; Li, H.; Fang, T.; Li, H.; Shi, Y.; Zhang, R.; Dong, X. A Multi-Medium Analysis of Human Health Risk of Toxic Elements in Rice-Crayfish System: A Case Study from Middle Reach of Yangtze River, China. Foods 2022, 11, 1160. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, J.; Gong, Z.; Yue, B.; Wang, X.; Manyande, A.; Du, H. Investigation of Bioaccumulation and Human Health Risk Assessment of Heavy Metals in Crayfish (Procambarus clarkii) Farming with a Rice-Crayfish-Based Coculture Breeding Modes. Foods 2022, 11, 261. [Google Scholar] [CrossRef]
- Zhang, D.; Fraser, M.A.; Huang, W.; Ge, C.; Wang, Y.; Zhang, C.; Guo, P. Microplastic pollution in water, sediment, and specific tissues of crayfish (Procambarus clarkii) within two different breeding modes in Jianli, Hubei province, China. Environ. Pollut 2021, 272, 115939. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Cornejo-Granados, F.; Gallardo-Becerra, L.; Leonardo-Reza, M.; Ochoa-Romo, J.P.; Ochoa-Leyva, A. A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 2018, 6, e5382. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Shi, G.; Luo, X.; Kang, J.; Su, J.; Wang, L. Effects of Growth Environment on Bacterial Community Diiversity of Procambarus clarkia. Sci. Technol. Food Ind. 2021, 42, 91–98. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Yu, Y.; Ni, J.; Xu, W.; Yan, Q. Composition of Gut Microbiota in the Gibel Carp (Carassius auratus gibelio) Varies with Host Development. Microb. Ecol. 2017, 74, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Weng, S.; He, J. Comparative analysis of the bacterial community compositions of the shrimp intestine, surrounding water and sediment. J. Appl. Microbiol. 2018, 125, 792–799. [Google Scholar] [CrossRef]
- Xiong, J.; Dai, W.; Qiu, Q.; Zhu, J.; Yang, W.; Li, C. Response of host-bacterial colonization in shrimp to developmental stage, environment and disease. Mol. Ecol. 2018, 27, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 734. [Google Scholar] [CrossRef]
- Eiler, A.; Bertilsson, S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ. Microbiol. 2004, 6, 1228–1243. [Google Scholar] [CrossRef]
- Holt, C.C.; Bass, D.; Stentiford, G.D.; van der Giezen, M. Understanding the role of the shrimp gut microbiome in health and disease. J. Invertebr. Pathol. 2021, 186, 107387. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 2017, 19, 1490–1501. [Google Scholar] [CrossRef]
- Shi, J.; Wu, X.; Huang, G.; Yang, H.; Lliang, Y.; Lyu, M.; Zeng, L.; Hu, D.; Huang, L.; Wang, R. Structural and Functional Characteristics of Intestinal Microbiota in Different Populations of Macrobrachium rosenbergii Larvae. Fish. Sci. 2023, 42, 674–681. [Google Scholar] [CrossRef]
- Li, M.; Shang, Q.; Li, G.; Wang, X.; Yu, G. Degradation of Marine Algae-Derived Carbohydrates by Bacteroidetes Isolated from Human Gut Microbiota. Mar. Drugs 2017, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.F.; Dixon, B.A. Enzyme production by obligate intestinal anaerobic bacteria isolated from oscars (Astronotus ocellatus), angelfish (Pterophyllum scalare) and southern flounder (Paralichthys lethostigma). Aquaculture 2003, 227, 417–426. [Google Scholar] [CrossRef]
- Van Hung, N.; De Schryver, P.; Dung, N.V.; Nevejan, N.; Bossier, P. Ralstonia eutropha, containing high poly-beta-hydroxybutyrate levels, regulates the immune response in mussel larvae challenged with Vibrio coralliilyticus. Fish Shellfish Immunol. 2019, 84, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Luo, J.; Du, H.; Xiong, W.; Qiao, X.; Wei, Q. Alterations in the gut microbiota in Chinese sturgeon (Acipenser sinensis) suffering from haemorrhagic septicaemia. Aquac. Res. 2021, 52, 6410–6419. [Google Scholar] [CrossRef]
- Yu, P.; Shan, H.; Cheng, Y.; Ma, J.; Wang, K.; Li, H. Translucent disease outbreak in Penaeus vannamei post-larva accompanies the imbalance of pond water and shrimp gut microbiota homeostasis. Aquac. Rep. 2022, 27, 101410. [Google Scholar] [CrossRef]
- Zhang, X.; Tao, Y.; Hu, J.; Liu, G.; Spanjers, H.; van Lier, J.B. Biomethanation and microbial community changes in a digester treating sludge from a brackish aquaculture recirculation system. Bioresour. Technol. 2016, 214, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.-J.; Amenyogbe, E.; Li, R.-X.; Zhang, J.-D.; Xie, R.-T.; Wang, Z.-L.; Chen, G.; Huang, J.-S.; Kumar, P. The Microflora Structure in the Digestive Tract, Culture Water, and Feed of Hybrid Grouper (Epinephelus fuscoguttatus♀ × E. polyphekadion♂) Cultured in an Outdoor Pond Based on a High-Throughput Sequencing Technique. Aquac. Res. 2023, 2023, 9923362. [Google Scholar] [CrossRef]
- Chapagain, P.; Walker, D.; Leeds, T.; Cleveland, B.M.; Salem, M. Distinct microbial assemblages associated with genetic selection for high- and low- muscle yield in rainbow trout. BMC Genom. 2020, 21, 820. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, H.; Zhang, D. Effects of different culture patterns on the intestinal microbiota of Litopenaeus vannamei. J. Fish. China 2021, 45, 221–234. [Google Scholar]
- Hou, Y.; Li, B.; Xu, G.; Li, D.; Zhang, C.; Jia, R.; Li, Q.; Zhu, J. Dynamic and Assembly of Benthic Bacterial Community in an Industrial-Scale In-Pond Raceway Recirculating Culture System. Front. Microbiol. 2021, 12, 797817. [Google Scholar] [CrossRef]
- Lu, L.; Yin, S.; Liu, X.; Zhang, W.; Gu, T.; Shen, Q.; Qiu, H. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef]
- Tseng, D.Y.; Ho, P.L.; Huang, S.Y.; Cheng, S.C.; Shiu, Y.L.; Chiu, C.S.; Liu, C.H. Enhancement of immunity and disease resistance in the white shrimp, Litopenaeus vannamei, by the probiotic, Bacillus subtilis E20. Fish Shellfish Immunol. 2009, 26, 339–344. [Google Scholar] [CrossRef]
- Zhou, M.; Hou, Y.; Jia, R.; Li, B.; Zhu, J. Effects of Bellamya purificata Cultivation at Different Stocking Densities on the Dynamics and Assembly of Bacterial Communities in Sediment. Biomolecules 2023, 13, 254. [Google Scholar] [CrossRef] [PubMed]
- de Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.M.; Henry, L.M.; Cornwallis, C.K.; Kiers, E.T.; West, S.A. The evolution of host-symbiont dependence. Nat. Commun. 2017, 8, 15973. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FX | SQ | XY | YZ | QJ | JL | HH | YY | CS | NX | |
|---|---|---|---|---|---|---|---|---|---|---|
| Nodes | 10 | 8 | 10 | 16 | 18 | 20 | 12 | 17 | 17 | 9 |
| Edges | 15 | 13 | 7 | 44 | 57 | 105 | 14 | 51 | 36 | 6 |
| Positive edge ratio | 80.00% | 53.85% | 71.43% | 52.27% | 59.65% | 46.67% | 100% | 49.02% | 66.67% | 100% |
| Negative edge ratio | 20.00% | 46.15% | 28.57% | 47.73% | 40.35% | 53.33% | 0% | 50.98% | 33.33% | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, C.; Jia, R.; Wang, Y.; Hou, Y.; Zhang, L.; Li, B.; Zhu, J. Comparison of Intestinal Bacteria of Procambarus clarkii Farmed in Various Rice Paddy Regions. Animals 2024, 14, 935. https://doi.org/10.3390/ani14060935
Ding C, Jia R, Wang Y, Hou Y, Zhang L, Li B, Zhu J. Comparison of Intestinal Bacteria of Procambarus clarkii Farmed in Various Rice Paddy Regions. Animals. 2024; 14(6):935. https://doi.org/10.3390/ani14060935
Chicago/Turabian StyleDing, Chonghang, Rui Jia, Yunfeng Wang, Yiran Hou, Liqiang Zhang, Bing Li, and Jian Zhu. 2024. "Comparison of Intestinal Bacteria of Procambarus clarkii Farmed in Various Rice Paddy Regions" Animals 14, no. 6: 935. https://doi.org/10.3390/ani14060935