
Genetic Diversity, Population Structure and Selection Signature in Begait Goats Revealed by Whole-Genome Sequencing
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Collection, DNA Extraction and Genomic Sequencing
2.2. Data Filtering, Clean Read Generation and Variant Discovery
2.3. Genetic Diversity, Population Differentiation and Population Structure Analysis
2.4. Genomic Linkage Disequilibrium (LD) and Effective Population Size (Ne)
2.5. Detection of the Genomic Regions under Selection
2.6. Functional Analysis of Candidate Genes under Selection
3. Results
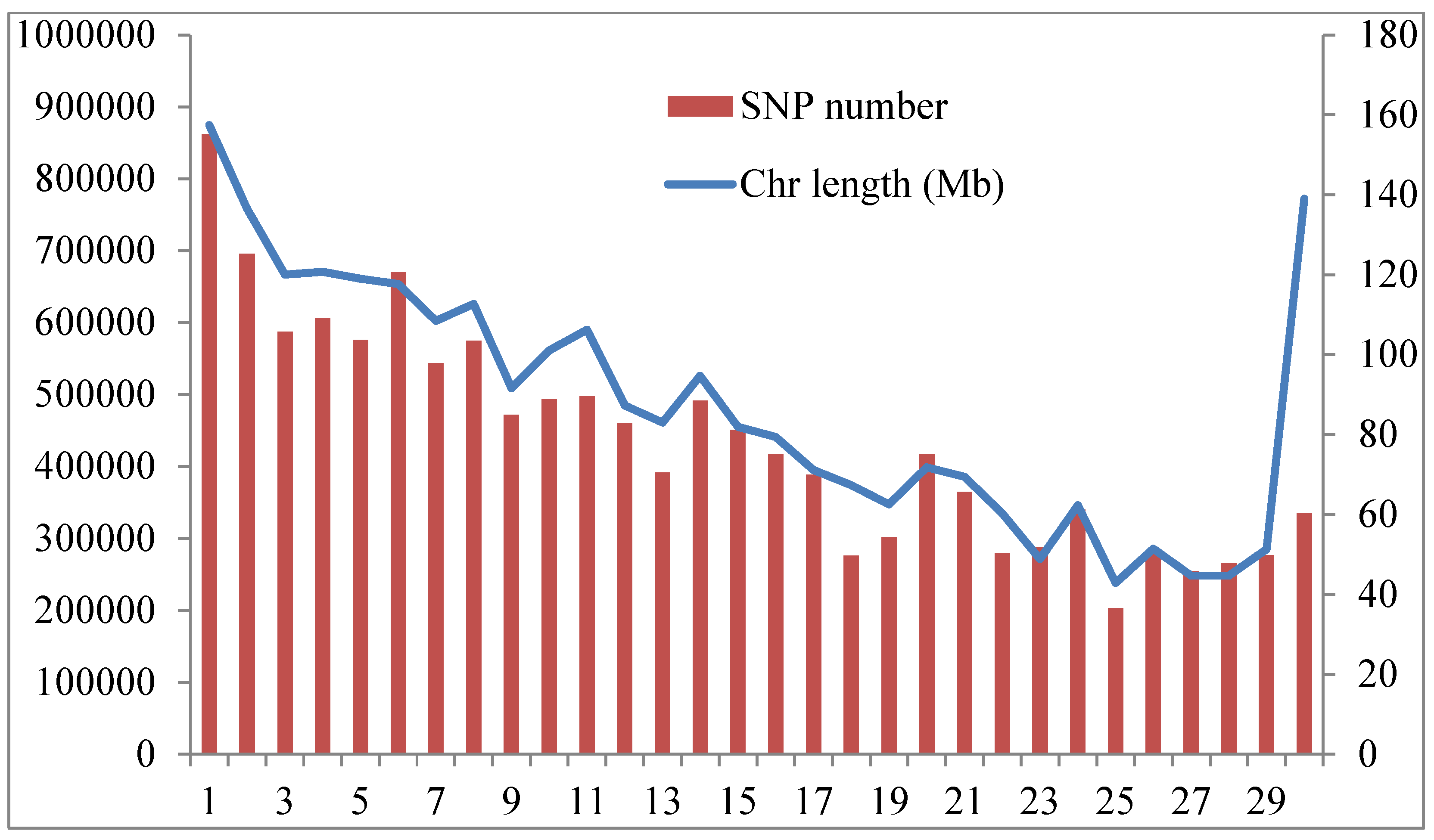
3.1. Overviews of Genetic Diversity
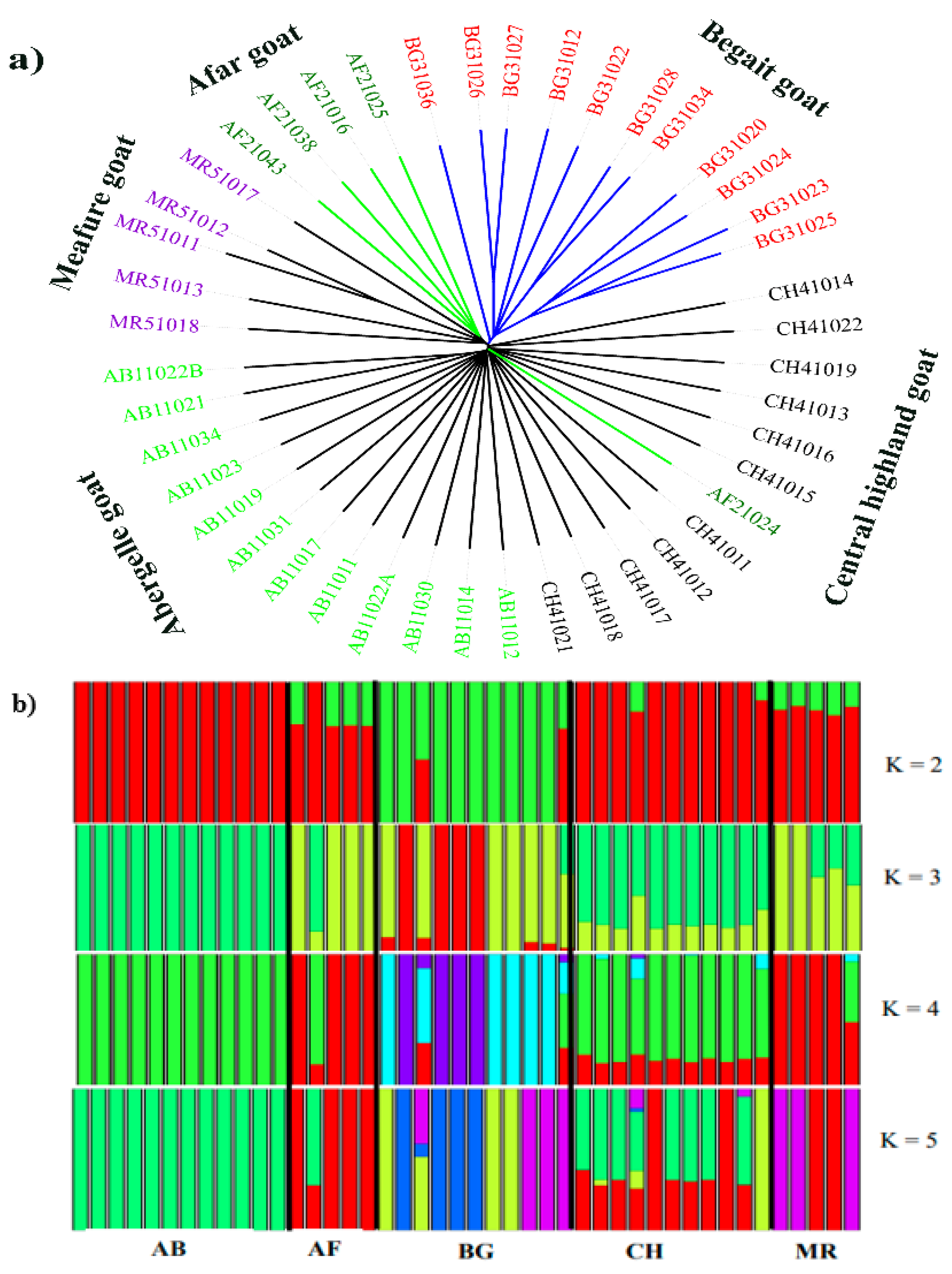
3.2. Population Structure Analysis
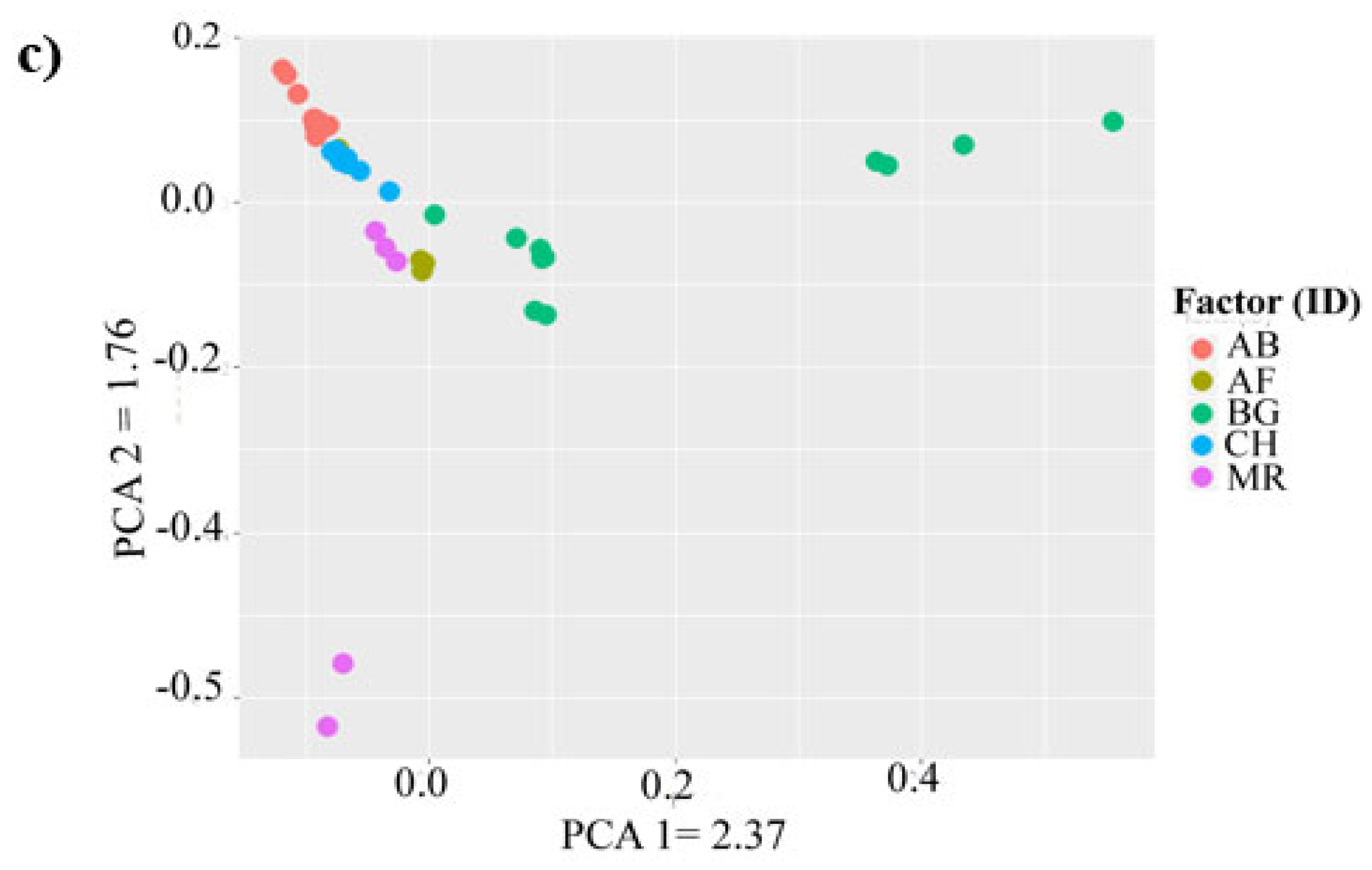
3.3. Genetic Relationship between Populations
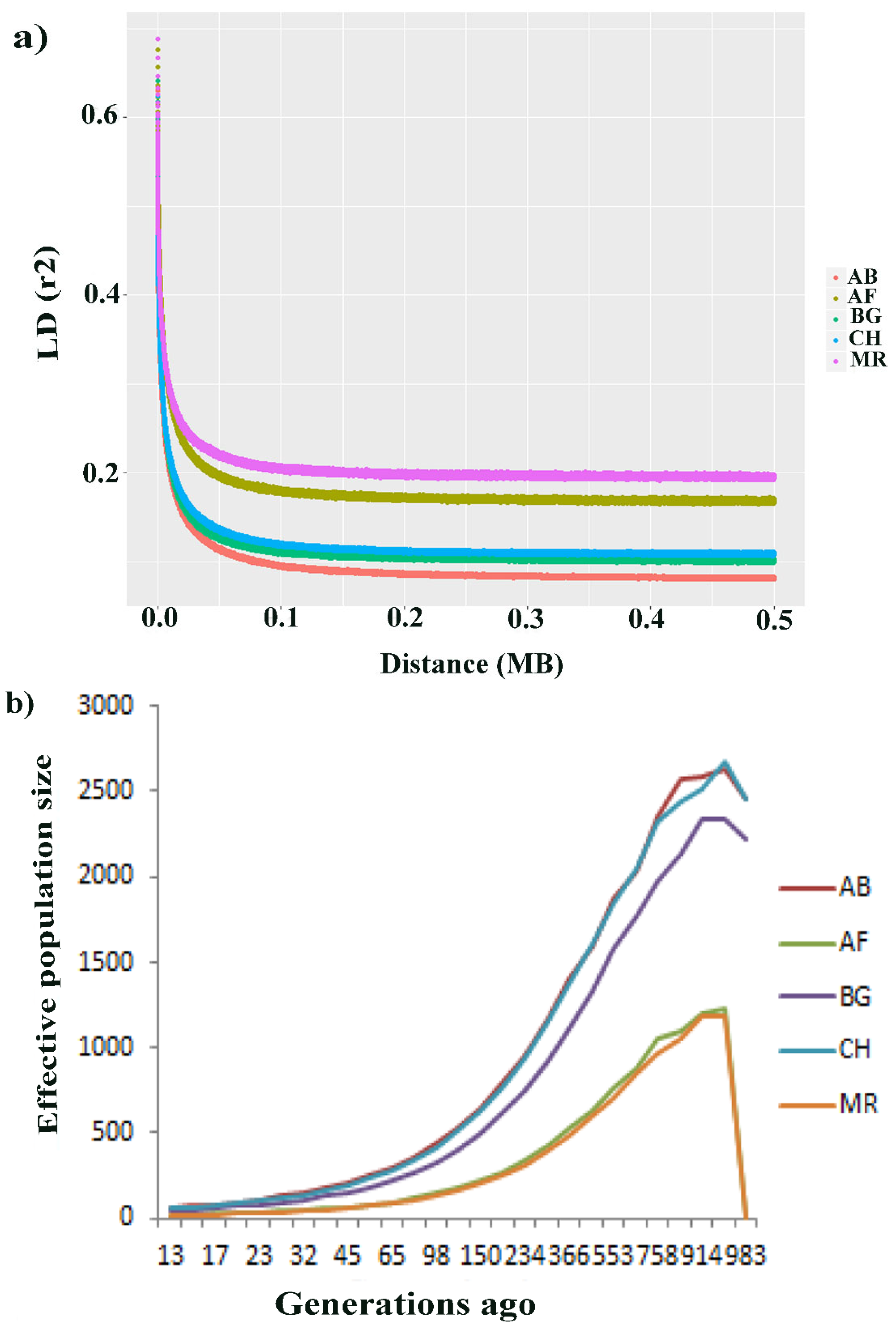
3.4. Genomic Linkage Disequilibrium (LD) and Effective Population Size (Ne)
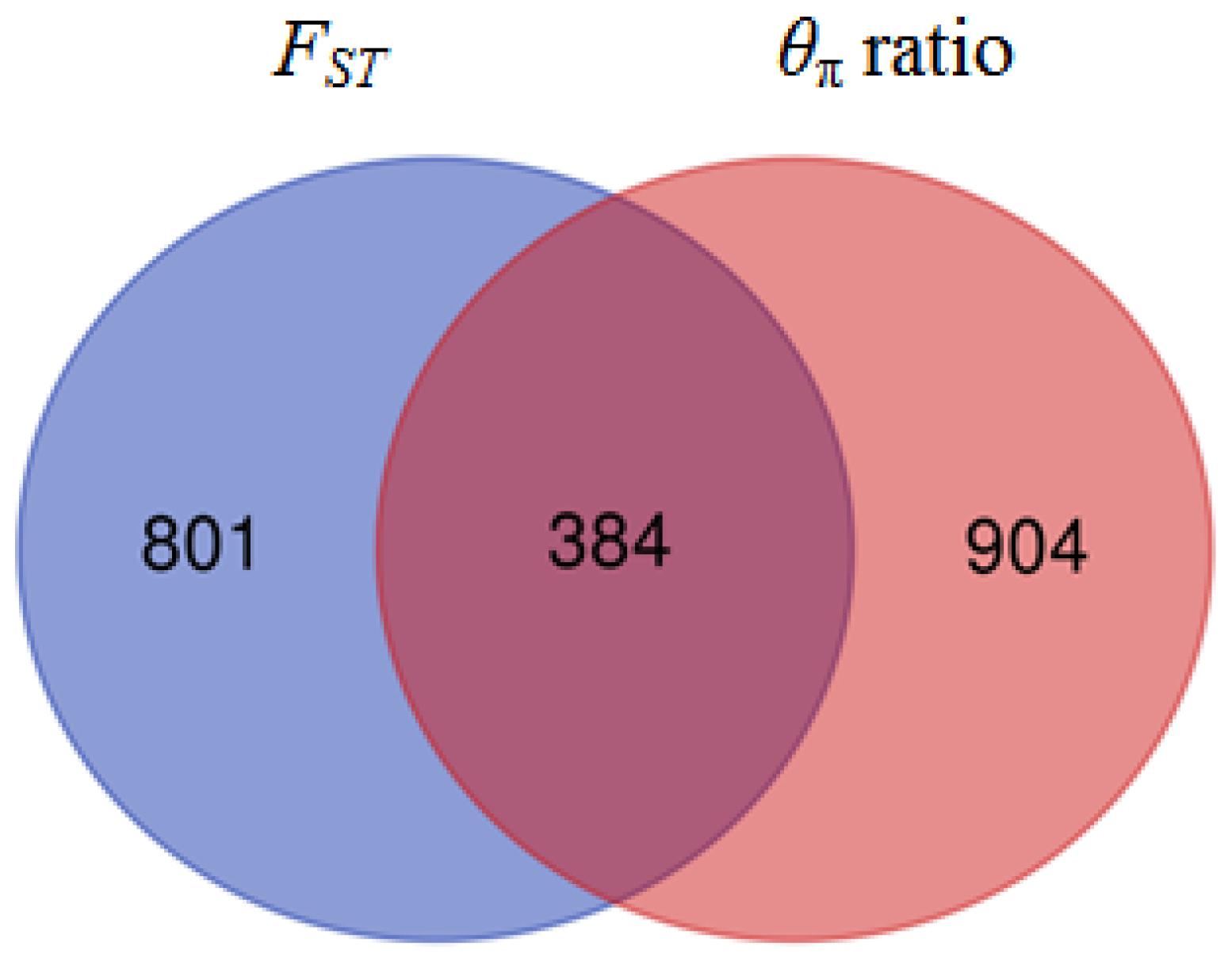
3.5. Detection of Selection Signatures
4. Discussion
4.1. Genetic Diversity Characteristics
4.2. Selection Signature and Identification of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Denoyelle, L.; Talouarn, E.; Bardou, P.; Colli, L.; Alberti, A.; Danchin, C.; Del Corvo, M.; Engelen, S.; Orvain, C.; Palhière, I.; et al. VarGoats Project: A Dataset of 1159 Whole-Genome Sequences to Dissect Capra hircus Global Diversity. Genet. Sel. Evol. 2021, 53, 86. [Google Scholar] [CrossRef] [PubMed]
- Berihulay, H.; Abied, A.; He, X.; Jiang, L.; Ma, Y. Adaptation Mechanisms of Small Ruminants to Environmental Heat Stress. Animals 2019, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, P.; Dai, X.; Asadollahpour Nanaei, H.; Fang, W.; Yang, Z.; Cai, Y.; Zheng, Z.; Wang, X.; Jiang, Y. A near Complete Genome for Goat Genetic and Genomic Research. Genet. Sel. Evol. 2021, 53, 74. [Google Scholar] [CrossRef]
- Aziz, M.A. Present Status of the World Goat Populations and Their Productivity. Lohmann Inf. 2010, 45, 42–52. [Google Scholar]
- CSA. Federal Democratic Republic of Ethiopia: Volume II Report on Livestock and Livestock Characteristics (Private Peasant Holdings); 2017. Available online: https://searchworks.stanford.edu/view/6509594 (accessed on 1 April 2021).
- Wan, X.; Jing, J.; Wang, D. Whole-Genome Selective Scans Detect Genes Associated with Important Phenotypic Traits in Goat (Capra hircu S). Front. Genet. 2023, 14, 1173017. [Google Scholar] [CrossRef] [PubMed]
- Mendeley Reference Manager. Available online: https://www.mendeley.com/reference-manager/reader/Attempting to use a disconnected port object/Attempting to use a disconnected port object/ (accessed on 15 November 2023).
- Abraham, H.; Gizaw, S.; Urge, M.; Abraham, H.; Gizaw, S.; Urge, M. Begait Goat Production Systems and Breeding Practices in Western Tigray, North Ethiopia. Open J. Anim. Sci. 2017, 7, 198–212. [Google Scholar] [CrossRef]
- Abraham, H. Identification of Breeding Objectives for Begait Goat in Western Tigray, North Ethiopia. Trop. Anim. Health Prod. 2018, 50, 1887–1892. [Google Scholar]
- Ghanatsaman, Z.A.; Mehrgardi, A.A.; Nanaei, H.A.; Esmailizadeh, A. Comparative Genomic Analysis Uncovers Candidate Genes Related with Milk Production and Adaptive Traits in Goat Breeds. Sci. Rep. 2023, 13, 8722. [Google Scholar] [CrossRef]
- Shi, H.; Li, T.; Su, M.; Wang, H.; Li, Q.; Lang, X.; Ma, Y. Whole Genome Sequencing Revealed Genetic Diversity, Population Structure, and Selective Signature of Panou Tibetan Sheep. BMC Genom. 2023, 24, 50. [Google Scholar] [CrossRef]
- Lai, F.N.; Zhai, H.L.; Cheng, M.; Ma, J.Y.; Cheng, S.F.; Ge, W.; Zhang, G.L.; Wang, J.J.; Zhang, R.Q.; Wang, X.; et al. Whole-Genome Scanning for the Litter Size Trait Associated Genes and SNPs under Selection in Dairy Goat (Capra hircus). Sci. Rep. 2016, 6, 38096. [Google Scholar] [CrossRef]
- Berihulay, H.; Li, Y.; Gebrekidan, B.; Gebreselassie, G.; Liu, X.; Jiang, L.; Ma, Y. Whole Genome Resequencing Reveals Selection Signatures Associated with Important Traits in Ethiopian Indigenous Goat Populations. Front. Genet. 2019, 10, 1190. [Google Scholar] [CrossRef]
- Li, X.; Su, R.; Wan, W.; Zhang, W.; Jiang, H.; Qiao, X.; Fan, Y.; Zhang, Y.; Wang, R.; Liu, Z.; et al. Identification of Selection Signals by Large-Scale Whole-Genome Resequencing of Cashmere Goats. Sci. Rep. 2017, 7, 15142. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A High-Performance Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Alexander, D.; Novembre, J.N.; Lange, K. Supplementary Material for Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Weir, B.S.; Clark Cockerham, C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A Tool to Estimate Trends in Recent Effective Population Size Trajectories Using Genome-Wide SNP Data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, A.B.; Rovadoscki, G.A.; Petrini, J.; Coutinho, L.L.; Morota, G.; Spangler, M.L.; Pinto, L.F.B.; Carvalho, G.G.P.; Mourão, G.B. Linkage Disequilibrium in Brazilian Santa Inês Breed, Ovis Aries. Sci. Rep. 2018, 8, 8851. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of Selection Signature Provides a Glimpse into Important Economic Traits in Goats (Capra hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef] [PubMed]
- Rahmatalla, S.A.; Arends, D.; Reissmann, M.; Ahmed, A.S.; Wimmers, K.; Reyer, H.; Brockmann, G.A. Whole Genome Population Genetics Analysis of Sudanese Goats Identifies Regions Harboring Genes Associated with Major Traits. BMC Genet. 2017, 18, 92. [Google Scholar] [CrossRef]
- Brito, L.F.; Kijas, J.W.; Ventura, R.V.; Sargolzaei, M.; Porto-Neto, L.R.; Cánovas, A.; Feng, Z.; Jafarikia, M.; Schenkel, F.S. Genetic Diversity and Signatures of Selection in Various Goat Breeds Revealed by Genome-Wide SNP Markers. BMC Genom. 2017, 18, 229. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yao, N.; Yang, M.; Liu, X.; Dong, K.; Zhao, Q.; Pu, Y.; He, X. Exome Sequencing Reveals Genetic Differentiation Due to High-Altitude Adaptation in the Tibetan Cashmere Goat (Capra hircus). BMC Genom. 2016, 17, 122. [Google Scholar] [CrossRef]
- Onzima, B.R.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Genome-Wide Characterization of Selection Signatures and Runs of Homozygosity in Ugandan Goat Breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef]
- Mdladla, K.; Dzomba, E.F.; Huson, H.J.; Muchadeyi, F.C. Population Genomic Structure and Linkage Disequilibrium Analysis of South African Goat Breeds Using Genome-Wide SNP Data. Anim. Genet. 2016, 47, 471–482. [Google Scholar]
- Lashmar, S.F.; Visser, C. SNP-Based Genetic Diversity of South African Commercial Dairy and Fiber Goat Breeds. Small Rumin. Res. 2016, 136, 65–71. [Google Scholar]
- Nicoloso, L.; Bomba, L.; Colli, L.; Negrini, R.; Milanesi, M.; Mazza, R.; Sechi, T.; Frattini, S.; Talenti, A.; Coizet, B.; et al. Genetic Diversity of Italian Goat Breeds Assessed with a Medium-Density SNP Chip. Genet. Sel. Evol. 2015, 9, 318. [Google Scholar] [CrossRef]
- Benjelloun, B.; Alberto, F.J.; Streeter, I.; Boyer, F.; Coissac, E.; Stucki, S.; BenBati, M.; Ibnelbachyr, M.; Chentouf, M.; Bechchari, A.; et al. Characterizing Neutral Genomic Diversity and Selection Signatures in Indigenous Populations of Moroccan Goats (Capra hircus) Using WGS Data. Front. Genet. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Onzima, R.B.; Upadhyay, M.R.; Mukiibi, R.; Kanis, E.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Genome-Wide Population Structure and Admixture Analysis Reveals Weak Differentiation among Ugandan Goat Breeds. Anim. Genet. 2018, 49, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Mekuriaw, G. Molecular Characterization of Ethiopian Indigenous Goat Populations: Genetic Diversity and Structure, Demographic Dynamics and Assessment of the Kisspeptin Gene Polymorphism. Ph.D. Thesis, Addis Ababa University, Addis Ababa, Ethiopia, 2016. [Google Scholar]
- Oliveira, J.C.V.; Ribeiro, M.N.; Rocha, L.L.; Gomes-filho, M.A.; Delgado, J.V. Genetic Relationships between Two Homologous Goat Breeds from Portugal and Brazil Assessed by Microsatellite Markers. Small Rumin. Res. 2010, 93, 79–87. [Google Scholar] [CrossRef]
- Visser, C.; Lashmar, S.F.; Van Marle-köster, E.; Poli, M.A. Genetic Diversity and Population Structure in South African, French and Argentinian Angora Goats from Genome-Wide SNP Data. PloS ONE 2016, 11, e0154353. [Google Scholar] [CrossRef] [PubMed]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective Population Size of an Indigenous Swiss Cattle Breed Estimated from Linkage Disequilibrium. J. Anim. Breed. Genet 2010, 127, 0931–2668. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, G.; Zeng, T.; Wei, C.; Zhang, L.; Wang, H.; Zhang, S.; Liu, R.; Liu, Z.; Du, L. Estimations of Genomic Linkage Disequilibrium and Effective Population Sizes in Three Sheep Populations. Livest. Sci. 2014, 170, 22–29. [Google Scholar] [CrossRef]
- Liu, S.; He, S.; Chen, L.; Li, W.; Di, J.; Liu, M. Estimates of Linkage Disequilibrium and Effective Population Sizes in Chinese Merino (Xinjiang Type) Sheep by Genome-Wide SNPs. Genes Genom. 2017, 39, 733–745. [Google Scholar] [CrossRef]
- Uimari, P.; Tapio, M. Extent of Linkage Disequilibrium and Effective Population Size in Finnish Landrace and Finnish Yorkshire Pig Breeds. J. Anim. Sci. 2011, 89, 609–614. [Google Scholar] [CrossRef]
- Waineina, R.W.; Ngeno, K.; Okeno, T.O.; Ilatsia, E.D. Genetic Diversity and Population Structure among Indigenous and Imported Goat Breeds in Kenya. Genet. Resour. 2021, 2, 25–35. [Google Scholar] [CrossRef]
- Berihulay, H.; Li, Y.; Liu, X.; Gebreselassie, G.; Islam, R.; Liu, W.; Jiang, L.; Ma, Y. Genetic Diversity and Population Structure in Multiple Chinese Goat Populations Using a SNP Panel. Anim. Genet. 2019, 50, 242–249. [Google Scholar] [CrossRef]
- Xiong, J.; Bao, J.; Hu, W.; Shang, M. Whole-Genome Resequencing Reveals Genetic Diversity and Selection Characteristics of Dairy Goat. Front. Genet. 2023, 13, 1044017. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-Genome Sequencing Reveals Selection Signatures Associated with Important Traits in Six Goat Breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar] [CrossRef]
- Yang, J.I.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M.; et al. Whole-Genome Sequencing of Native Sheep Provides Insights into Rapid Adaptations to Extreme Environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef]
- Terefe, E.; Belay, G.; Tijjani, A.; Han, J.; Hanotte, O. Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments. Diversity 2023, 15, 540. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, M.; Cheng, H.; Fan, W.; Yuan, Z.; Gao, Q.; Xu, Y.; Guo, Z.; Zhang, Y.; Hu, J.; et al. An Intercross Population Study Reveals Genes Associated with Body Size and Plumage Color in Ducks. Nat. Commun. 2018, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Berhane, G.; Eik, L.O. Effect of Vetch (Vicia sativa) Hay Supplementation on Performance of Begait and Abergelle Goats in Northern Ethiopia I. Milk Yield and Composition. Small Rumin. Res. 2006, 64, 225–232. [Google Scholar] [CrossRef]
- Mwacharo, J.M.; Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.A.; Rothschild, M.F. Genomic Footprints of Dryland Stress Adaptation in Egyptian Fat-Tail Sheep and Their Divergence from East African and Western Asia Cohorts. Sci. Rep. 2017, 7, 17647. [Google Scholar] [CrossRef]
- Kim, J.; Lee, T.; Kim, T.-H.; Lee, K.-T.; Kim, H. An Integrated Approach of Comparative Genomics and Heritability Analysis of Pig and Human on Obesity Trait: Evidence for Candidate Genes on Human Chromosome 2. BMC Genom. 2012, 13, 711. [Google Scholar] [CrossRef]
- Oxburgh, L. Control of the Bone Morphogenetic Protein 7 Gene in Developmental and Adult Life. Curr. Genom. 2009, 10, 223–230. [Google Scholar] [CrossRef]
- Gould, S.E.; Day, M.; Jones, S.S.; Doral, H. Bmp-7 Regulates Chemokine, Cytokine, and Hemodynamic Gene Expression in Proximal Tubule Cells. Kidney Int. 2002, 61, 51–60. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone Morphogenetic Proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Park, L. Linkage Disequilibrium Decay and Past Population History in the Human Genome. PLoS ONE 2012, 7, e46603. [Google Scholar] [CrossRef]
- Seo, M.; Kim, K.; Yoon, J.; Jeong, J.Y.; Lee, H.J.; Cho, S.; Kim, H. RNA-Seq Analysis for Detecting Quantitative Trait-Associated Genes. Sci. Rep. 2016, 6, 24375. [Google Scholar] [CrossRef]
- Taneera, J.; Fadista, J.; Ahlqvist, E.; Atac, D.; Ottosson-Laakso, E.; Wollheim, C.B.; Groop, L. Identification of Novel Genes for Glucose Metabolism Based upon Expression Pattern in Human Islets and Effect on Insulin Secretion and Glycemia. Hum. Mol. Genet. 2014, 24, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Jevsinek Skok, D.; Kunej, T.; Kovac, M.; Malovrh, S.; Potocnik, K.; Petric, N.; Zgur, S.; Dovc, P.; Horvat, S. FTO Gene Variants Are Associated with Growth and Carcass Traits in Cattle. Anim. Genet. 2016, 47, 219–222. [Google Scholar] [CrossRef]
- Dina, C.; Meyre, D.; Gallina, S.; Durand, E.; Körner, A.; Jacobson, P.; Carlsson, L.M.S.; Kiess, W.; Vatin, V.; Lecoeur, C.; et al. Variation in FTO Contributes to Childhood Obesity and Severe Adult Obesity. Nat. Genet. 2007, 39, 724–726. [Google Scholar] [CrossRef]
- Schessl, J.; Bach, E.; Rost, S.; Feldkirchner, S.; Kubny, C.; Müller, S.; Hanisch, F.G.; Kress, W.; Schoser, B. Novel Recessive Myotilin Mutation Causes Severe Myofibrillar Myopathy. Neurogenetics 2014, 15, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Adoligbe, C.M.; Zan, L.; Farougou, S. P3017 Functional Analysis and Association Studies of Bovine MYOT Gene with Meat Quality. J. Anim. Sci. 2009, 94, 59–60. [Google Scholar]
- Le Provost, F.; Cassy, S.; Hayes, H.; Martin, P. Structure and Expression of Goat GLYCAM1 Gene: Lactogenic-Dependent Expression in Ruminant Mammary Gland and Interspecies Conservation of the Proximal Promoter. Gene 2003, 313, 83–89. [Google Scholar] [CrossRef]
- Groenen, M.A.M.; Dijkhof, R.J.M.; Poel, J.J. van der Characterization of a GlyCAMl-like Gene (Glycosylation-Depegndent Cell Adhesion Molecule 1) Which Is Highly and Specifically Expressed in the Lactating Bovine Mammary Gland. Gene 1995, 158, 189–195. [Google Scholar] [CrossRef]
- Liu, W.; Wang, J.; Li, Q.; Ju, Z.; Huang, J.; Wang, H.; Liu, S.; Li, J.; Zhong, J.; Wang, C. Correlation Analysis between Three Novel SNPs of the Src Gene in Bovine and Milk Production Traits. Mol. Biol. Rep. 2010, 37, 3771–3777. [Google Scholar] [CrossRef]
- Watkin, H.; Richert, M.M.; Lewis, A.; Terrell, K.; McManaman, J.P.; Anderson, S.M. Lactation Failure in Src Knockout Mice Is Due to Impaired Secretory Activation. BMC Dev. Biol. 2008, 8, 6. [Google Scholar] [CrossRef]
- Daramola, J.O.; Adeloye, A.A. Physiological Adaptation to the Humid Tropics with Special Reference to the West African Dwarf (WAD) Goat. Trop. Anim. Health Prod. 2009, 41, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Poscic, N.; Montanari, T.; Andrea, M.D.; Licastro, D.; Pilla, F.; Ajmone-marsan, P.; Minuti, A.; Sgorlon, S. Breed and Adaptive Response Modulate Bovine Peripheral Blood Cells’ Transcriptome. J. Anim. Sci. Biotechnol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; Steri, R.; et al. Signatures of Selection and Environmental Adaptation across the Goat Genome Post-Domestication. Genet. Sel. Evol. GSE 2018, 50, 57. [Google Scholar] [CrossRef] [PubMed]
- Kapila, N.; Sharma, A.; Kishore, A.; Sodhi, M.; Tripathi, P.K.; Mohanty, A.K.; Mukesh, M. Impact of Heat Stress on Cellular and Transcriptional Adaptation of Mammary Epithelial Cells in Riverine Buffalo (Bubalus bubalis). PLoS ONE 2016, 11, e0157237. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple Genomic Signatures of Selection in Goats and Sheep Indigenous to a Hot Arid Environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Garrett-sinha, L.A. Review of Ets1 Structure, Function, and Roles in Immunity. Cell Mol. Life Sci. 2014, 70, 3375–3390. [Google Scholar] [CrossRef] [PubMed]
- Légaré, C.; Akintayo, A.; Blondin, P.; Calvo, E.; Sullivan, R. Impact of Male Fertility Status on the Transcriptome of the Bovine Epididymis. Mol. Hum. Reprod. 2017, 23, 355–369. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Goat Breeds | Location | Altitude (m.a.s.l) | GPS Coordinator | Classification of Agro-Ecologies | |
|---|---|---|---|---|---|---|
| Latitude | Longitude | |||||
| 1 | Begait | Tigray region (Western zone) | 560–1849 | 13°14′–14°27′ N | 36°27′ E–37°32′ E | low altitude |
| 2 | Aberegalle | Tigray (Central zone) | 1200–1500 | 13°19′60.00′ N | 38°49′59.99″ E | low altitude |
| 3 | Afar | Afar (Abeala) | 100–1500 | 13°21′23.05′ N | 39°45′27.10″ E | low altitude |
| 4 | Central highlands | Tigray (Central zone) | 1500–2300 | 14′0715.92′ N | 38°43′24.13′ E | mid altitude |
| 5 | Meafure | Tigray (Eastern zone) | 1500–2300 | 11°23′–11°44′ N | 38°20′–38°44′ E | mid altitude |
| Population | n | SNPs | PN | HO | HE | MAF < 0.05 | FIS |
|---|---|---|---|---|---|---|---|
| Abergelle | 12 | 11,137,576 | 82.69 | 32.2 | 34.5 | 0.222 | 0.03 |
| Afar | 5 | 10,760,581 | 78.62 | 22.8 | 26.6 | 0.214 | −0.04 |
| Begait | 11 | 10,833,847 | 85.42 | 35.2 | 40.2 | 0.221 | 0.01 |
| Central highland | 11 | 12,229,657 | 82.52 | 30.7 | 30.8 | 0.224 | 0.04 |
| Meafure | 5 | 10,749,996 | 78.58 | 26.3 | 29.7 | 0.223 | −0.18 |
| Breed Name | AB | AF | BG | CH | MR |
|---|---|---|---|---|---|
| AB | 0.107 | 0.065 | 0.151 | 0.078 | |
| AF | 0.014 | 0.105 | 0.036 | 0.123 | |
| BG | 0.182 | 0.019 | 0.15 | 0.078 | |
| CH | 0.003 | 0.006 | 0.027 | 0.16 | |
| MR | 0.019 | 0.007 | 0.028 | 0.011 |
| Traits | Chr. | Position (Mb) | Candidate Genes | Z (FST) | log2 (θπ, Ratio) |
|---|---|---|---|---|---|
| Defensive mechanism | 13 | 60.30–60.50 | DEFB116, DEFB119, DEFB124 | 6.37 | 0.55 |
| Melanogenesis | 13 | 59.70–59.90 | SLC52A3 | 3.65 | 2.38 |
| 13 | 25.20–25.40 | SLC6A2 | 4.11 | 0.31 | |
| Growth and milk quality traits | 5 | 25.20–25.40 | GLYCAM1, PPP1R1A | 4.36 | 1.37 |
| 18 | 23.90–24.10 | FTO | 3.83 | 0.32 | |
| 13 | 65.80–66.00 | SRC | 2.62 | 0.36 | |
| Heat tolerance | 23 | 38.60–38.80 | MAPK13, MAPK14 | 2.45 | 0.75 |
| Category | Term | p Value | Genes |
|---|---|---|---|
| KEGG_PATHWAY | hsa04261: Adrenergic signaling in cardiomyocytes | 0.001 | AGTR1, MAPK13, MAPK14, BCL2, PPP1R1A, CACNB1, CAMK2D, SCN7A, CACNA2D2 |
| KEGG_PATHWAY | hsa04912: GnRH signaling pathway | 0.01 | MAPK13, MAPK14, CAMK2D, PRKCD, MMP2, SRC |
| KEGG_PATHWAY | hsa05152: Tuberculosis | 0.02 | MAPK13, MAPK14, BCL2, IL12A, CAMK2D, ATP6V1H, EEA1, SRC |
| KEGG_PATHWAY | hsa05014: Amyotrophic lateral sclerosis (ALS) | 0.04 | GRIN2B, MAPK13, MAPK14, BCL2 |
| KEGG_PATHWAY | hsa05169: Epstein–Barr virus infection | 0.04 | ITGAL, PSMD12, MAPK13, MAPK14, BCL2, PSMD2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebreselase, H.B.; Nigussie, H.; Wang, C.; Luo, C. Genetic Diversity, Population Structure and Selection Signature in Begait Goats Revealed by Whole-Genome Sequencing. Animals 2024, 14, 307. https://doi.org/10.3390/ani14020307
Gebreselase HB, Nigussie H, Wang C, Luo C. Genetic Diversity, Population Structure and Selection Signature in Begait Goats Revealed by Whole-Genome Sequencing. Animals. 2024; 14(2):307. https://doi.org/10.3390/ani14020307
Chicago/Turabian StyleGebreselase, Haile Berihulay, Hailemichael Nigussie, Changfa Wang, and Chenglong Luo. 2024. "Genetic Diversity, Population Structure and Selection Signature in Begait Goats Revealed by Whole-Genome Sequencing" Animals 14, no. 2: 307. https://doi.org/10.3390/ani14020307