

Deep Sequencing of Porcine Reproductive and Respiratory Syndrome Virus ORF7: A Promising Tool for Diagnostics and Epidemiologic Surveillance
 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strains and Serum Samples
2.2. RNA Isolation and RT-qPCR
2.3. Design of ORF7 Primers
2.4. Two-Step RT-PCR-Based Amplicon Library Preparation
2.5. Analysis of NGS Data
3. Results
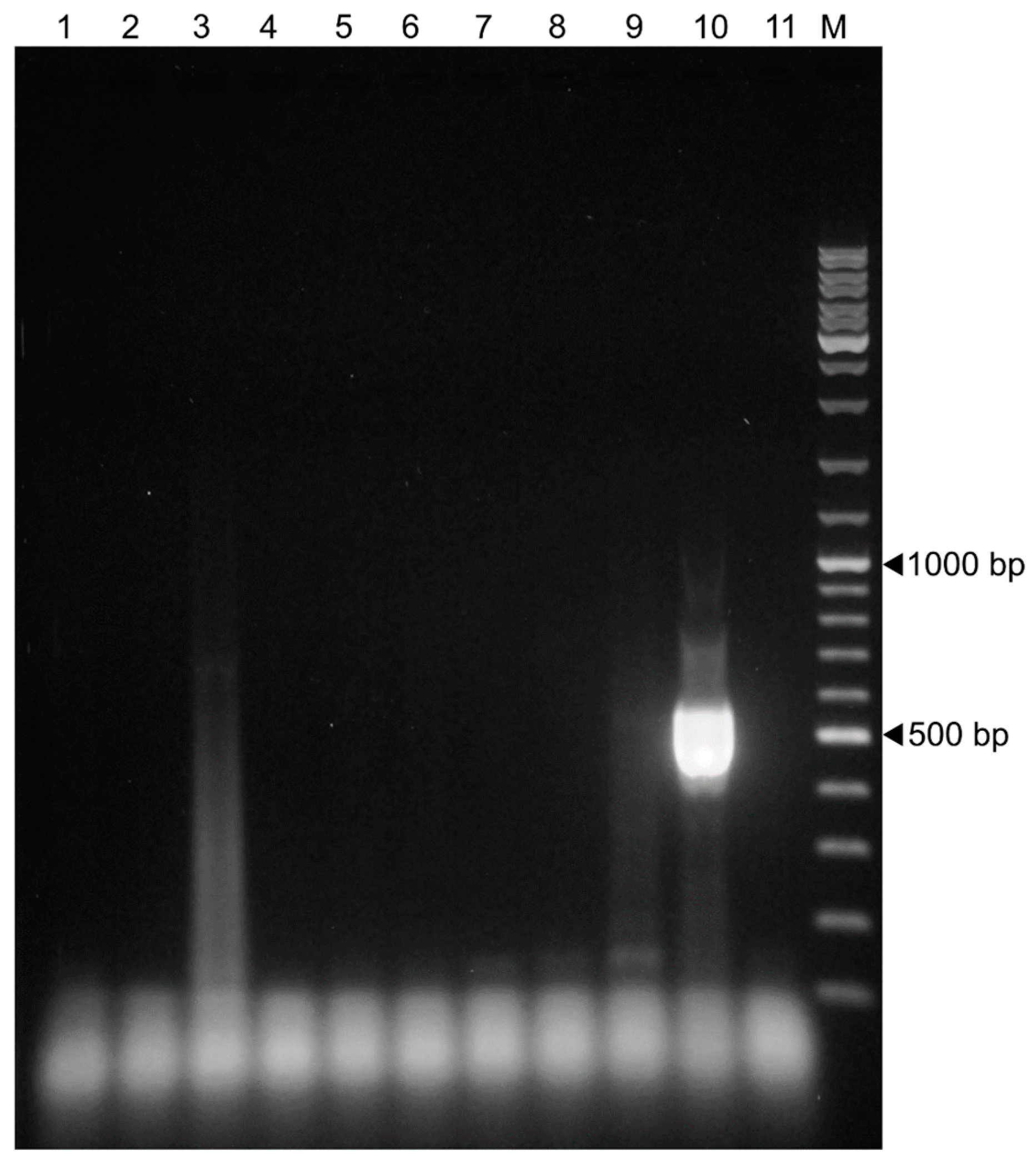
3.1. Assay Specificity
3.2. Assay Sensitivity
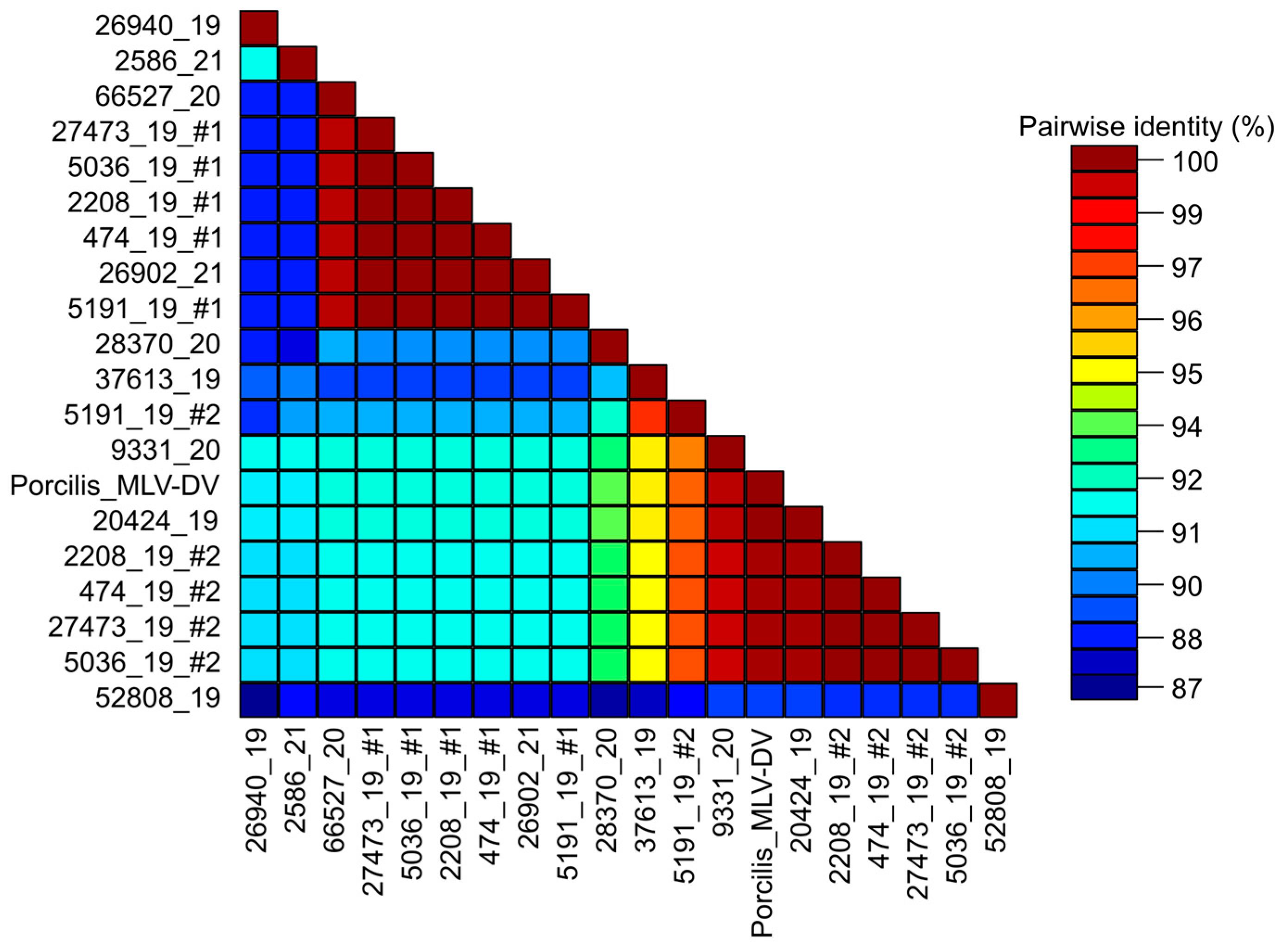
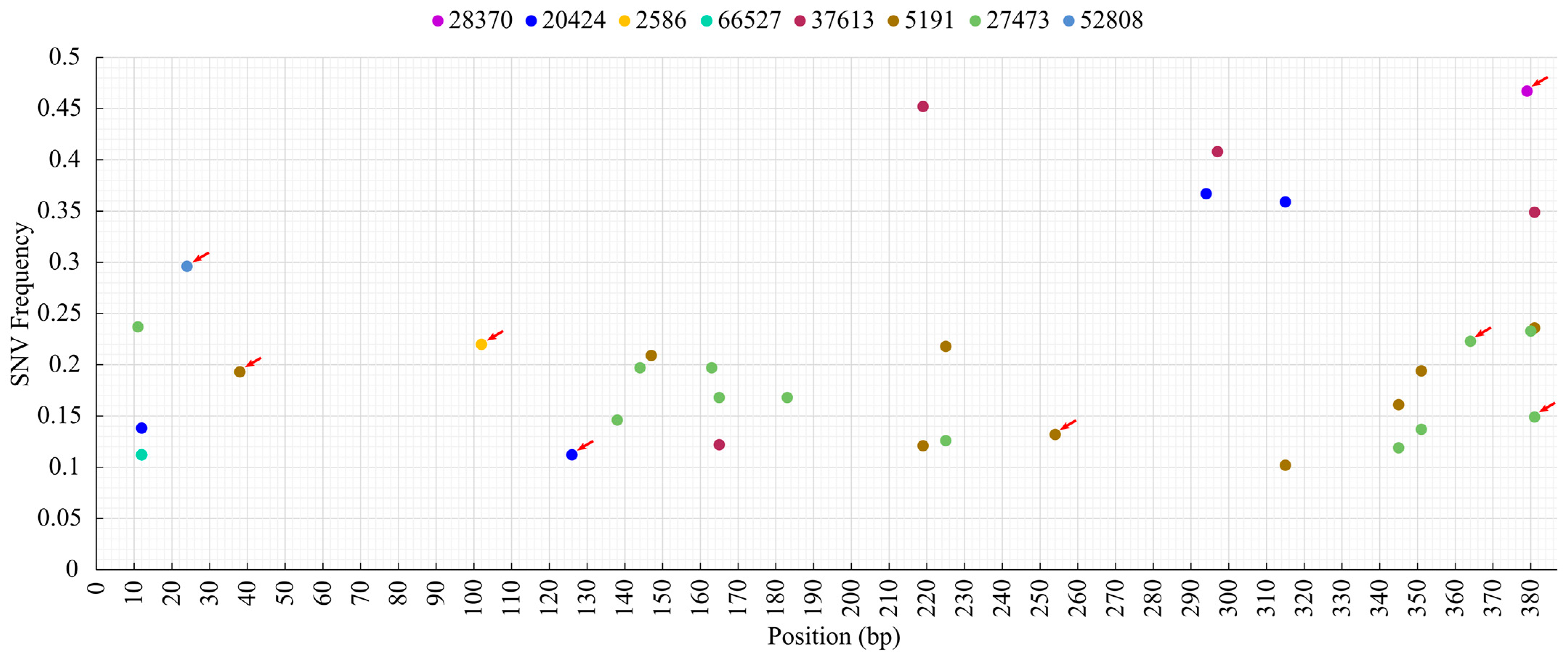
3.3. Analysis of Sequence Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allende, R.; Lewis, T.L.; Lu, Z.; Rock, D.L.; Kutish, G.F.; Ali, A.; Doster, A.R.; Osorio, F.A. North American and European Porcine Reproductive and Respiratory Syndrome Viruses Differ in Non-Structural Protein Coding Regions. J. Gen. Virol. 1999, 80, 307–315. [Google Scholar] [CrossRef]
- Murtaugh, M.P.; Stadejek, T.; Abrahante, J.E.; Lam, T.T.Y.; Leung, F.C.C. The Ever-Expanding Diversity of Porcine Reproductive and Respiratory Syndrome Virus. Virus Res. 2010, 154, 18–30. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.-H.; Li, J.; Wong, L.T.-W.; Yip, C.-W.; Jiang, J.-W.; et al. Phylogeny-Based Evolutionary, Demographical, and Geographical Dissection of North American Type 2 Porcine Reproductive and Respiratory Syndrome Viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Kin-Hei Hui, R.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Chi-Ching Leung, F. Molecular Epidemiology of PRRSV: A Phylogenetic Perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Balka, G.; Podgórska, K.; Brar, M.S.; Bálint, Á.; Cadar, D.; Celer, V.; Dénes, L.; Dirbakova, Z.; Jedryczko, A.; Márton, L.; et al. Genetic Diversity of PRRSV 1 in Central Eastern Europe in 1994–2014: Origin and Evolution of the Virus in the Region. Sci. Rep. 2018, 8, 7811. [Google Scholar] [CrossRef]
- Cui, X.; Xia, D.; Huang, X.; Sun, Y.; Shi, M.; Zhang, J.; Li, G.; Yang, Y.; Wang, H.; Cai, X.; et al. Analysis of Recombinant Characteristics Based on 949 PRRSV-2 Genomic Sequences Obtained from 1991 to 2021 Shows That Viral Multiplication Ability Contributes to Dominant Recombination. Microbiol. Spectr. 2022, 10, e02934-22. [Google Scholar] [CrossRef]
- Holtkamp, D.J.; Polson, D.D.; Torremorell, M.; Morrison, B.; Classen, D.M.; Becton, L.; Henry, S.; Rodibaugh, M.T.; Rowland, R.R.; Snelson, H.; et al. Terminology for Classifying Swine Herds by Porcine Reproductive and Respiratory Syndrome Virus Status. J. Swine Health Prod. 2011, 19, 44–56. [Google Scholar] [CrossRef]
- Berton, P.; Normand, V.; Martineau, G.-P.; Bouchet, F.; Lebret, A.; Waret-Szkuta, A. Evaluation of Porcine Reproductive and Respiratory Syndrome Stabilization Protocols in 23 French Farrow-to-Finish Farms Located in a High-Density Swine Area. Porc. Health Manag. 2017, 3, 11. [Google Scholar] [CrossRef]
- Alvarez, J.; Valdes-Donoso, P.; Tousignant, S.; Alkhamis, M.; Morrison, R.; Perez, A. Novel Analytic Tools for the Study of Porcine Reproductive and Respiratory Syndrome Virus (PRRSv) in Endemic Settings: Lessons Learned in the U.S. Porc. Health Manag. 2016, 2, 3. [Google Scholar] [CrossRef]
- Perez, A.M.; Davies, P.R.; Goodell, C.K.; Holtkamp, D.J.; Mondaca-Fernández, E.; Poljak, Z.; Tousignant, S.J.; Valdes-Donoso, P.; Zimmerman, J.J.; Morrison, R.B. Lessons Learned and Knowledge Gaps about the Epidemiology and Control of Porcine Reproductive and Respiratory Syndrome Virus in North America. J. Am. Vet. Med. Assoc. 2015, 246, 1304–1317. [Google Scholar] [CrossRef]
- Murtaugh, M.P. Use and Interpretation of Sequencing in PRRSV Control Programs. In Proceedings of the Allen D. Leman Swine Conference, St. Paul, MN, USA, 18–21 September 2012; Volume 39, pp. 49–55. [Google Scholar]
- Paploski, I.A.D.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder, D.C.; VanderWaal, K. Temporal Dynamics of Co-Circulating Lineages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10, 2486. [Google Scholar] [CrossRef] [PubMed]
- Bálint, Á.; Molnár, T.; Kecskeméti, S.; Kulcsár, G.; Soós, T.; Szabó, P.M.; Kaszab, E.; Fornyos, K.; Zádori, Z.; Bányai, K.; et al. Genetic Variability of PRRSV Vaccine Strains Used in the National Eradication Programme, Hungary. Vaccines 2021, 9, 849. [Google Scholar] [CrossRef]
- Kikuti, M.; Sanhueza, J.; Vilalta, C.; Paploski, I.A.D.; VanderWaal, K.; Corzo, C.A. Porcine Reproductive and Respiratory Syndrome Virus 2 (PRRSV-2) Genetic Diversity and Occurrence of Wild Type and Vaccine-like Strains in the United States Swine Industry. PLoS ONE 2021, 16, e0259531. [Google Scholar] [CrossRef] [PubMed]
- Rupasinghe, R.; Lee, K.; Liu, X.; Gauger, P.C.; Zhang, J.; Martínez-López, B. Molecular Evolution of Porcine Reproductive and Respiratory Syndrome Virus Field Strains from Two Swine Production Systems in the Midwestern United States from 2001 to 2020. Microbiol. Spectr. 2022, 10, e02634-21. [Google Scholar] [CrossRef]
- Jakab, S.; Kaszab, E.; Marton, S.; Bányai, K.; Bálint, Á.; Nemes, I.; Szabó, I. Genetic Diversity of Imported PRRSV-2 Strains, 2005–2020, Hungary. Front. Vet. Sci. 2022, 9, 1581. [Google Scholar] [CrossRef]
- Cheng, T.Y.; Campler, M.R.; Schroeder, D.C.; Yang, M.; Mor, S.K.; Ferreira, J.B.; Arruda, A.G. Detection of Multiple Lineages of PRRSV in Breeding and Growing Swine Farms. Front. Vet. Sci. 2022, 9, 884733. [Google Scholar] [CrossRef]
- Nilubol, D.; Tripipat, T.; Hoonsuwan, T.; Tipsombatboon, P.; Piriyapongsa, J. Genetic Diversity of the ORF5 Gene of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Genotypes I and II in Thailand. Arch. Virol. 2013, 158, 943–953. [Google Scholar] [CrossRef]
- Greiser-Wilke, I.; Fiebig, K.; Drexler, C.; grosse Beilage, E. Genetic Diversity of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in Selected Herds in a Pig-Dense Region of North-Western Germany. Vet. Microbiol. 2010, 143, 213–223. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; Santin, M. Next Generation Amplicon Sequencing Improves Detection of Blastocystis Mixed Subtype Infections. Infect. Genet. Evol. 2019, 73, 119–125. [Google Scholar] [CrossRef]
- Quer, J.; Gregori, J.; Rodríguez-Frias, F.; Buti, M.; Madejon, A.; Perez-del-Pulgar, S.; Garcia-Cehic, D.; Casillas, R.; Blasi, M.; Homs, M.; et al. High-Resolution Hepatitis C Virus Subtyping Using NS5B Deep Sequencing and Phylogeny, an Alternative to Current Methods. J. Clin. Microbiol. 2015, 53, 219–226. [Google Scholar] [CrossRef]
- Ramakrishnan, M.A.; Tu, Z.J.; Singh, S.; Chockalingam, A.K.; Gramer, M.R.; Wang, P.; Goyal, S.M.; Yang, M.; Halvorson, D.A.; Sreevatsan, S. The Feasibility of Using High Resolution Genome Sequencing of Influenza A Viruses to Detect Mixed Infections and Quasispecies. PLoS ONE 2009, 4, e7105. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.; Ip, C.L.C.; Badhan, A.; Christiansen, M.T.; Adamson, W.; Ansari, M.A.; Bibby, D.; Breuer, J.; Brown, A.; Bowden, R.; et al. Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes. J. Clin. Microbiol. 2016, 54, 2470–2484. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, A.L.; Sreenu, V.B.; Wilkie, G.; Gunson, R.; Templeton, K.; Leitch, E.C.M. Prevalence of Mixed Genotype Hepatitis C Virus Infections in the UK as Determined by Genotype-specific PCR and Deep Sequencing. J. Viral Hepat. 2018, 25, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Sikkema-Raddatz, B.; Johansson, L.F.; de Boer, E.N.; Almomani, R.; Boven, L.G.; van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; Sijmons, R.H.; Jongbloed, J.D.H.; et al. Targeted Next-Generation Sequencing Can Replace Sanger Sequencing in Clinical Diagnostics. Hum. Mutat. 2013, 34, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, G.; Zeller, M.; Li, G.; Zhang, J.; Gauger, P.; Linhares, D.C.L. Implementing a User-friendly Format to Analyze PRRSV Next-generation Sequencing Results and Associating Breeding Herd Production Performance with Number of PRRSV Strains and Recombination Events. Transbound. Emerg. Dis. 2022, 69, e2214–e2229. [Google Scholar] [CrossRef]
- Kim, S.-C.; Moon, S.-H.; Jeong, C.-G.; Park, G.-S.; Park, J.-Y.; Jeoung, H.-Y.; Shin, G.-E.; Ko, M.-K.; Kim, S.-H.; Lee, K.-K.; et al. Whole-Genome Sequencing and Genetic Characteristics of Representative Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Isolates in Korea. Virol. J. 2022, 19, 66. [Google Scholar] [CrossRef]
- Brar, M.S.; Shi, M.; Hui, R.K.-H.; Leung, F.C.-C. Genomic Evolution of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Isolates Revealed by Deep Sequencing. PLoS ONE 2014, 9, e88807. [Google Scholar] [CrossRef]
- Kvisgaard, L.K.; Hjulsager, C.K.; Fahnøe, U.; Breum, S.Ø.; Ait-Ali, T.; Larsen, L.E. A Fast and Robust Method for Full Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 and Type 2. J. Virol. Methods 2013, 193, 697–705. [Google Scholar] [CrossRef]
- Rajkhowa, T.K.; Thanga, L.; Hauhnar, L.; Zodinpui, D.; Subbiah, M. Molecular Detection and Characterization of Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus from a Natural Outbreak in Wild Pigs, Mizoram, India. Transbound. Emerg. Dis. 2022, 69, e288–e298. [Google Scholar] [CrossRef]
- Lu, Z.H.; Archibald, A.L.; Ait-Ali, T. Beyond the Whole Genome Consensus: Unravelling of PRRSV Phylogenomics Using next Generation Sequencing Technologies. Virus Res. 2014, 194, 167–174. [Google Scholar] [CrossRef]
- Yu, F.; Yan, Y.; Shi, M.; Liu, H.-Z.; Zhang, H.-L.; Yang, Y.-B.; Huang, X.-Y.; Gauger, P.C.; Zhang, J.; Zhang, Y.-H.; et al. Phylogenetics, Genomic Recombination, and NSP2 Polymorphic Patterns of Porcine Reproductive and Respiratory Syndrome Virus in China and the United States in 2014–2018. J. Virol. 2020, 94, 1813–1832. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zheng, Z.; Cao, X.; Wang, Z.; Xu, Z.; Gao, H.; Liu, J.; Xu, S.; Lin, J.; Chen, S.; et al. Whole Genome Sequencing of Clinical Specimens Reveals the Genomic Diversity of Porcine Reproductive and Respiratory Syndrome Viruses Emerging in China. Transbound. Emerg. Dis. 2022, 69, e2530–e2540. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kim, S.-C.; Kim, H.-J.; Jeong, C.-G.; Park, G.-S.; Choi, J.-S.; Kim, W.-I. Insight into the Economic Effects of a Severe Korean PRRSV1 Outbreak in a Farrow-to-Nursery Farm. Animals 2022, 12, 3024. [Google Scholar] [CrossRef] [PubMed]
- Mötz, M.; Stadler, J.; Kreutzmann, H.; Ladinig, A.; Lamp, B.; Auer, A.; Riedel, C.; Rümenapf, T. A Conserved Stem-Loop Structure within ORF5 Is a Frequent Recombination Hotspot for Porcine Reproductive and Respiratory Syndrome Virus 1 (PRRSV-1) with a Particular Modified Live Virus (MLV) Strain. Viruses 2023, 15, 258. [Google Scholar] [CrossRef]
- Lu, Z.H.; Wang, X.; Wilson, A.D.; Dorey-Robinson, D.L.W.; Archibald, A.L.; Ait-Ali, T.; Frossard, J.-P. Quasispecies Evolution of the Prototypical Genotype 1 Porcine Reproductive and Respiratory Syndrome Virus Early during In Vivo Infection Is Rapid and Tissue Specific. Arch. Virol. 2017, 162, 2203–2210. [Google Scholar] [CrossRef]
- Rowland, R.R.R.; Doerksen, T.; Lu, A.; Sheahan, M.; Lunney, J.; Dekkers, J.; Palinski, R.M. Effect of the Host Genotype at a Porcine Reproductive and Respiratory Syndrome (PRRS) Resistance Marker on Evolution of the Modified-Live PRRS Vaccine Virus in Pigs. Virus Res. 2022, 316, 198809. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Xia, X.-Q.; Chen, Q.; Bade, S.A.; Yoon, K.-J.; Harmon, K.M.; Gauger, P.C.; Main, R.G.; Li, G. High-Throughput Whole Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus from Cell Culture Materials and Clinical Specimens Using next-Generation Sequencing Technology. J. Vet. Diagn. Investig. 2017, 29, 41–50. [Google Scholar] [CrossRef]
- Gagnon, C.A.; Lalonde, C.; Provost, C. Porcine Reproductive and Respiratory Syndrome Virus Whole-Genome Sequencing Efficacy with Field Clinical Samples Using a Poly(A)-Tail Viral Genome Purification Method. J. Vet. Diagn. Investig. 2021, 33, 216–226. [Google Scholar] [CrossRef]
- Vandenbussche, F.; Mathijs, E.; Tignon, M.; Vandersmissen, T.; Cay, A.B. WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study. Viruses 2021, 13, 2419. [Google Scholar] [CrossRef]
- Lalonde, C.; Provost, C.; Gagnon, C.A. Whole-Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus from Field Clinical Samples Improves the Genomic Surveillance of the Virus. J. Clin. Microbiol. 2020, 58, e00097-20. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Rapid, Unbiased PRRSV Strain Detection Using MinION Direct RNA Sequencing and Bioinformatics Tools. Viruses 2019, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Jakab, S.; Marton, S.; Szabó, I.; Kecskeméti, S.; Bálint, Á.; Bányai, K. Preliminary Observations Concerning the Integration of Amplicon Deep Sequencing in the PRRS Eradication Program. Hung. Vet. J. 2022, 144, 115–128. [Google Scholar]
- Chen, N.; Trible, B.R.; Kerrigan, M.A.; Tian, K.; Rowland, R.R.R. ORF5 of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Is a Target of Diversifying Selection as Infection Progresses from Acute Infection to Virus Rebound. Infect. Genet. Evol. 2016, 40, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S. Veterinary Infection Biology: Molecular Diagnostics and High-Throughput Strategies; Cunha, M.V., Inácio, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1247, ISBN 978-1-4939-2003-7. [Google Scholar]
- Kumar, D. Next-Generation Sequencing as Diagnostic Tool in Veterinary Research. J. Anim. Res. 2019, 9, 500078. [Google Scholar] [CrossRef]
- Van Borm, S.; Wang, J.; Granberg, F.; Colling, A. Next-Generation Sequencing Workflows in Veterinary Infection Biology: Towards Validation and Quality Assurance. Rev. Sci. Tech. Off. Int. Epiz. 2016, 35, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Suminda, G.G.D.; Bhandari, S.; Won, Y.; Goutam, U.; Kanth Pulicherla, K.; Son, Y.-O.; Ghosh, M. High-Throughput Sequencing Technologies in the Detection of Livestock Pathogens, Diagnosis, and Zoonotic Surveillance. Comput. Struct. Biotechnol. J. 2022, 20, 5378–5392. [Google Scholar] [CrossRef]
- Belák, S.; Karlsson, O.E.; Leijon, M.; Grangerb, F. High-Throughput Sequencing in Veterinary Infection Biology and Diagnostics. Rev. Sci. Tech. Off. Int. Epiz. 2013, 32, 893–915. [Google Scholar] [CrossRef]
- Kaszab, E.; Doszpoly, A.; Lanave, G.; Verma, A.; Bányai, K.; Malik, Y.S.; Marton, S. Metagenomics Revealing New Virus Species in Farm and Pet Animals and Aquaculture. In Genomics and Biotechnological Advances in Veterinary, Poultry, and Fisheries; Elsevier: Amsterdam, The Netherlands, 2019; p. 564. ISBN 9780128163528. [Google Scholar]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Eriksson, C.E.; Ruprecht, J.; Levi, T. More Affordable and Effective Noninvasive Single Nucleotide Polymorphism Genotyping Using High-throughput Amplicon Sequencing. Mol. Ecol. Resour. 2020, 20, 1505–1516. [Google Scholar] [CrossRef]
- Bybee, S.M.; Bracken-Grissom, H.; Haynes, B.D.; Hermansen, R.A.; Byers, R.L.; Clement, M.J.; Udall, J.A.; Wilcox, E.R.; Crandall, K.A. Targeted Amplicon Sequencing (TAS): A Scalable Next-Gen Approach to Multilocus, Multitaxa Phylogenetics. Genome Biol. Evol. 2011, 3, 1312–1323. [Google Scholar] [CrossRef]
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; et al. Multiple Approaches for Massively Parallel Sequencing of SARS-CoV-2 Genomes Directly from Clinical Samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Gohl, D.M.; Garbe, J.; Grady, P.; Daniel, J.; Watson, R.H.B.; Auch, B.; Nelson, A.; Yohe, S.; Beckman, K.B. A Rapid, Cost-Effective Tailed Amplicon Method for Sequencing SARS-CoV-2. BMC Genom. 2020, 21, 863. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.B.; Humphrys, M.S.; Robinson, C.K.; Settles, M.L.; Ott, S.; Fu, L.; Yang, H.; Gajer, P.; He, X.; McComb, E.; et al. Ultrahigh-Throughput Multiplexing and Sequencing of >500-Base-Pair Amplicon Regions on the Illumina HiSeq 2500 Platform. mSystems 2019, 4, e00029-19. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Kegler, K.; Posthaus, H.; Perreten, V.; Rodriguez-Campos, S. Amplicon Sequencing of Bacterial Microbiota in Abortion Material from Cattle. Vet. Res. 2017, 48, 64. [Google Scholar] [CrossRef] [PubMed]
- Avramenko, R.W.; Redman, E.M.; Lewis, R.; Yazwinski, T.A.; Wasmuth, J.D.; Gilleard, J.S. Exploring the Gastrointestinal “Nemabiome”: Deep Amplicon Sequencing to Quantify the Species Composition of Parasitic Nematode Communities. PLoS ONE 2015, 10, e0143559. [Google Scholar] [CrossRef]
- Johnston, D.; Earley, B.; Cormican, P.; Murray, G.; Kenny, D.A.; Waters, S.M.; McGee, M.; Kelly, A.K.; McCabe, M.S. Illumina MiSeq 16S Amplicon Sequence Analysis of Bovine Respiratory Disease Associated Bacteria in Lung and Mediastinal Lymph Node Tissue. BMC Vet. Res. 2017, 13, 118. [Google Scholar] [CrossRef]
- Wolf-Jäckel, G.A.; Strube, M.L.; Schou, K.K.; Schnee, C.; Agerholm, J.S.; Jensen, T.K. Bovine Abortions Revisited—Enhancing Abortion Diagnostics by 16S rDNA Amplicon Sequencing and Fluorescence In Situ Hybridization. Front. Vet. Sci. 2021, 8, 623666. [Google Scholar] [CrossRef]
- Lowman, M.E.; Tipton, C.D.; Labordère, A.L.; Brown, J.A. Equine Sinusitis Aetiology Is Linked to Sinus Microbiome by Amplicon Sequencing. Equine Vet. J. 2023, 55, 798–807. [Google Scholar] [CrossRef]
- Quer, J.; Colomer-Castell, S.; Campos, C.; Andrés, C.; Piñana, M.; Cortese, M.F.; González-Sánchez, A.; Garcia-Cehic, D.; Ibáñez, M.; Pumarola, T.; et al. Next-Generation Sequencing for Confronting Virus Pandemics. Viruses 2022, 14, 600. [Google Scholar] [CrossRef]
- Kvisgaard, L.K.; Larsen, L.E.; Hjulsager, C.K.; Bøtner, A.; Rathkjen, P.H.; Heegaard, P.M.H.; Bisgaard, N.P.; Nielsen, J.; Hansen, M.S. Genetic and Biological Characterization of a Porcine Reproductive and Respiratory Syndrome Virus 2 (PRRSV-2) Causing Significant Clinical Disease in the Field. Vet. Microbiol. 2017, 211, 74–83. [Google Scholar] [CrossRef]
- Sinn, L.J.; Klingler, E.; Lamp, B.; Brunthaler, R.; Weissenböck, H.; Rümenapf, T.; Ladinig, A. Emergence of a Virulent Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) 1 Strain in Lower Austria. Porc. Health Manag. 2016, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Kvisgaard, L.K.; Hjulsager, C.K.; Brar, M.S.; Leung, F.C.C.; Larsen, L.E. Genetic Dissection of Complete Genomes of Type 2 PRRS Viruses Isolated in Denmark over a Period of 15 Years. Vet. Microbiol. 2013, 167, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Stadejek, T.; Oleksiewicz, M.B.; Scherbakov, A.V.; Timina, A.M.; Krabbe, J.S.; Chabros, K.; Potapchuk, D. Definition of Subtypes in the European Genotype of Porcine Reproductive and Respiratory Syndrome Virus: Nucleocapsid Characteristics and Geographical Distribution in Europe. Arch. Virol. 2008, 153, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Dortmans, J.C.F.M.; Buter, G.J.; Dijkman, R.; Houben, M.; Duinhof, T.F. Molecular Characterization of Type 1 Porcine Reproductive and Respiratory Syndrome Viruses (PRRSV) Isolated in the Netherlands from 2014 to 2016. PLoS ONE 2019, 14, e0218481. [Google Scholar] [CrossRef] [PubMed]
- Clilverd, H.; Martín-Valls, G.; Li, Y.; Martín, M.; Cortey, M.; Mateu, E. Infection Dynamics, Transmission, and Evolution after an Outbreak of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2023, 14, 1109881. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ct Value | ORF5 Clade | Filtered Reads | Sequencing Depth (X) | Average SNV (%) | |
|---|---|---|---|---|---|---|
| Major Variant | Minor Variant * | |||||
| 26940_19 | 20.9 | 1A | 502,722 | 149,864 | - | - |
| 2208_19 | 27.8 | 1E | 22,306 | 1029 | 75 | 25 |
| 28370_20 | 26.5 | 1E | 109,948 | 41,137 | - | - |
| 20424_19 | 21.5 | 1F | 154,252 | 38,505 | - | - |
| 26902_21 | 19.1 | 1G | 93,512 | 38,471 | - | - |
| 2586_21 | 16.5 | 1G | 33,762 | 4351 | - | - |
| 474_19 | 22.3 | 1G | 15,406 | 305 | 78 | 22 |
| 66527_20 | 22.0 | Porcilis | 172,782 | 67,857 | - | - |
| 9331_20 | 24.4 | Porcilis | 105,896 | 13,271 | - | - |
| 37613_19 | 22.6 | Porcilis-like | 31,070 | 6245 | - | - |
| 5191_19 | 27.8 | Reprocyc | 35,600 | 5846 | 53 | 47 |
| 27473_19 | 29.1 | Spanish | 5330 | 1605 | 58 | 42 |
| 5036_19 ° | 25.8 | Spanish | 898 | 202 | 62 | 38 |
| 30156_20 | 31.6 | 3D | - | - | - | - |
| 52808_19 | 26.5 | 3F | 6856 | 939 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakab, S.; Bali, K.; Freytag, C.; Pataki, A.; Fehér, E.; Halas, M.; Jerzsele, Á.; Szabó, I.; Szarka, K.; Bálint, Á.; et al. Deep Sequencing of Porcine Reproductive and Respiratory Syndrome Virus ORF7: A Promising Tool for Diagnostics and Epidemiologic Surveillance. Animals 2023, 13, 3223. https://doi.org/10.3390/ani13203223
Jakab S, Bali K, Freytag C, Pataki A, Fehér E, Halas M, Jerzsele Á, Szabó I, Szarka K, Bálint Á, et al. Deep Sequencing of Porcine Reproductive and Respiratory Syndrome Virus ORF7: A Promising Tool for Diagnostics and Epidemiologic Surveillance. Animals. 2023; 13(20):3223. https://doi.org/10.3390/ani13203223
Chicago/Turabian StyleJakab, Szilvia, Krisztina Bali, Csongor Freytag, Anna Pataki, Enikő Fehér, Máté Halas, Ákos Jerzsele, István Szabó, Krisztina Szarka, Ádám Bálint, and et al. 2023. "Deep Sequencing of Porcine Reproductive and Respiratory Syndrome Virus ORF7: A Promising Tool for Diagnostics and Epidemiologic Surveillance" Animals 13, no. 20: 3223. https://doi.org/10.3390/ani13203223