
PorcineAI-Enhancer: Prediction of Pig Enhancer Sequences Using Convolutional Neural Networks

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. High Confidence Sequence Acquisition
2.3. Sequence Coding Method
2.4. Sequence Analysis
2.5. CNN Model Architecture
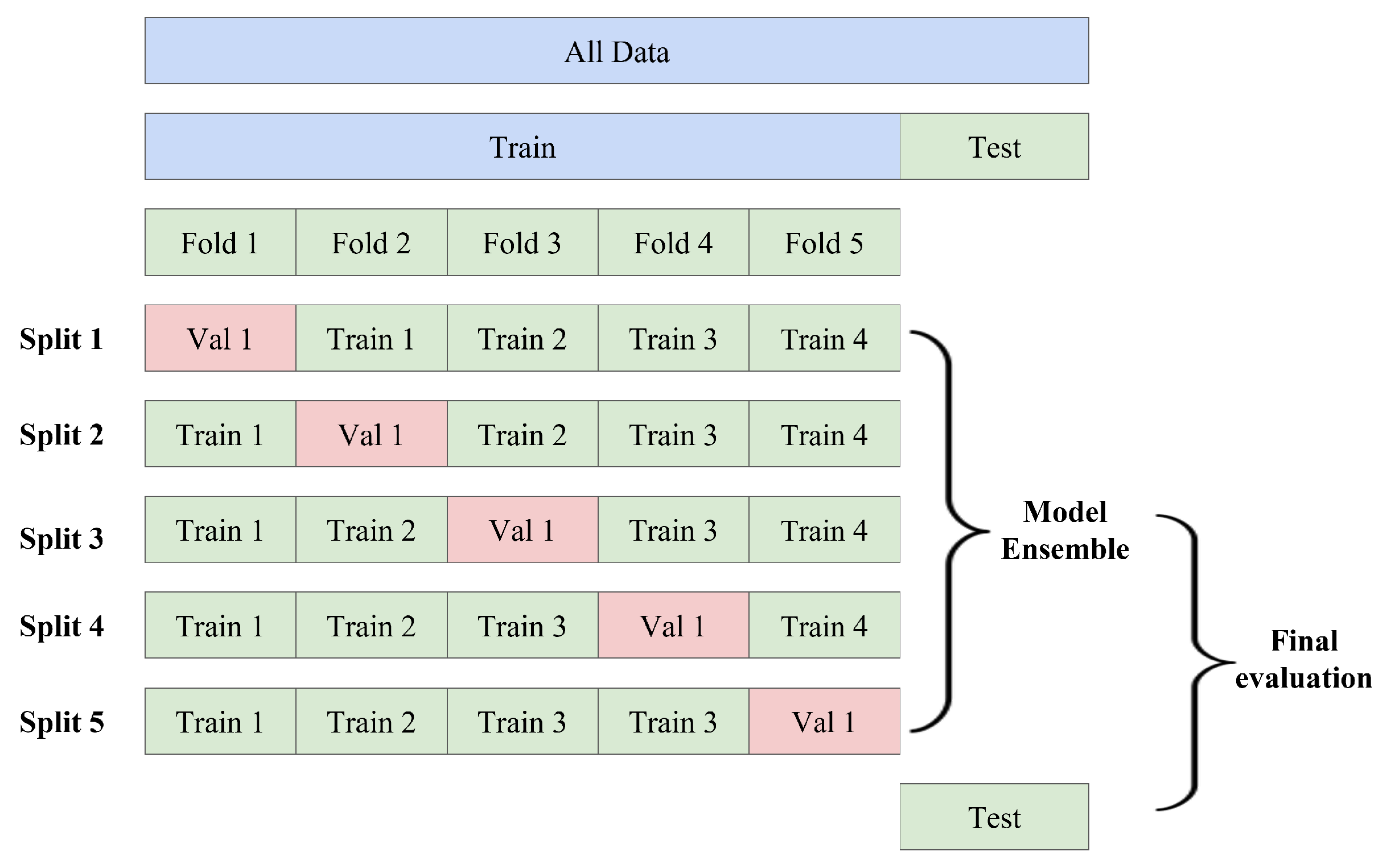
2.6. K-Fold Cross-Validation
3. Results
3.1. Sequence Analysis
3.2. PorcineAI-Enhancer Model Training
3.3. Performance of the PorcineAI-Enhancer Model
3.4. Comparison with Ensemble Model
3.5. Comparison with Existing State-of-the-Art Methods
3.6. Model Performance on Tissue-Specific Enhancers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmitz, R.J.; Grotewold, E.; Stam, M. Cis-regulatory sequences in plants: Their importance, discovery, and future challenges. Plant Cell 2021, 34, 718–741. [Google Scholar] [CrossRef]
- Beagan, J.A.; Pastuzyn, E.D.; Fernandez, L.R.; Guo, M.H.; Feng, K.; Titus, K.R.; Chandrashekar, H.; Shepherd, J.D.; Phillips-Cremins, J.E. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci. 2020, 23, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8, 592164. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Higgs, D.R. Enhancer–promoter interactions and transcription. Nat. Genet. 2020, 52, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Enhancers: The abundance and function of regulatory sequences beyond promoters. Dev. Biol. 2010, 339, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2012, 44, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, Y.; Wu, F.; Gui, Y.; Chen, X.; Wang, Y.; Wang, X.; Gui, Y.; Li, Q. Functional assessment of heart-specific enhancers by integrating ChIP-seq data. Pediatr. Res. 2022, 92, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Kang, R.; Ding, R.; Kang, W.; Zhang, Y.; Liu, W.; Huang, Q.; Meng, J.; Guo, Z. Genome-wide Identification and Characterization of Enhancers Across 10 Human Tissues. Int. J. Biol. Sci. 2018, 14, 1321–1332. [Google Scholar] [CrossRef]
- Droog, M.; Nevedomskaya, E.; Dackus, G.M.; Fles, R.; Kim, Y.; Hollema, H.; Mourits, M.J.; Nederlof, P.M.; van Boven, H.H.; Linn, S.C.; et al. Estrogen receptor α wields treatment-specific enhancers between morphologically similar endometrial tumors. Proc. Natl. Acad. Sci. USA 2017, 114, E1316–E1325. [Google Scholar] [CrossRef]
- Farley, E.K.; Olson, K.M.; Zhang, W.; Rokhsar, D.S.; Levine, M.S. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. USA 2016, 113, 6508–6513. [Google Scholar] [CrossRef]
- Liao, M.; Zhao, J.P.; Tian, J.; Zheng, C.H. iEnhancer-DCLA: Using the original sequence to identify enhancers and their strength based on a deep learning framework. BMC Bioinf. 2022, 23, 480. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Bristow, J.; Pennacchio, L.A. Enhancer identification through comparative genomics. Semin. Cell Dev. Biol. 2007, 18, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Erwin, G.D.; Oksenberg, N.; Truty, R.M.; Kostka, D.; Murphy, K.K.; Ahituv, N.; Pollard, K.S.; Capra, J.A. Integrating Diverse Datasets Improves Developmental Enhancer Prediction. PLoS Comput. Biol. 2014, 10, e1003677. [Google Scholar] [CrossRef]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- Bissonnette, P. Extraction and Identification of Frequent Sequential Patterns in Transcription Factor Binding Site Organization of Enhancers. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2014. [Google Scholar]
- Robey, A.; Hassani, H.; Pappas, G.J. Model-based robust deep learning: Generalizing to natural, out-of-distribution data. arXiv 2020, arXiv:2005.10247. [Google Scholar]
- Huang, Z.; Johnson, T.S.; Han, Z.; Helm, B.; Cao, S.; Zhang, C.; Salama, P.; Rizkalla, M.; Yu, C.Y.; Cheng, J.; et al. Deep learning-based cancer survival prognosis from RNA-seq data: Approaches and evaluations. BMC Med. Genom. 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.K.; Pradhan, C.; Das, H. Performance evaluation of different machine learning methods and deep-learning based convolutional neural network for health decision making. In Nature Inspired Computing for Data Science; Springer International Publishing: Cham, Switzerland, 2019; pp. 201–212. [Google Scholar] [CrossRef]
- Liu, D. Connecting Low-Level Image Processing and High-Level Vision via Deep Learning. In Proceedings of the 27th International Joint Conference on Artificial Intelligence (IJCAI-ECAI-18), Stockholm, Sweden, 13–19 July 2018. [Google Scholar] [CrossRef]
- Andrew, W.; Greatwood, C.; Burghardt, T. Visual localisation and individual identification of Holstein Friesian Cattle via deep learning. In Proceedings of the IEEE International Conference on Computer Vision Workshops, Venice, Italy, 22–29 October 2017; pp. 2850–2859. [Google Scholar] [CrossRef]
- Luongo, F.; Hakim, R.; Nguyen, J.H.; Anandkumar, A.; Hung, A.J. Deep learning-based computer vision to recognize and classify suturing gestures in robot-assisted surgery. Surgery 2021, 169, 1240–1244. [Google Scholar] [CrossRef]
- Song, Z. English speech recognition based on deep learning with multiple features. Computing 2019, 102, 663–682. [Google Scholar] [CrossRef]
- Trong, T.N.; Hautamäki, V.; Lee, K.A. Deep Language: A comprehensive deep learning approach to end-to-end language recognition. In Proceedings of the Speaker and Language Recognition Workshop (Odyssey 2016), Bilbao, Spain, 21–24 June 2016. [Google Scholar] [CrossRef]
- Cai, J.; Liu, Y. Research on English pronunciation training based on intelligent speech recognition. Int. J. Speech Technol. 2018, 21, 633–640. [Google Scholar] [CrossRef]
- Liu, B.; Fang, L.; Long, R.; Lan, X.; Chou, K.C. iEnhancer-2L: A two-layer predictor for identifying enhancers and their strength by pseudok-tuple nucleotide composition. Bioinformatics 2016, 32, 362–369. [Google Scholar] [CrossRef]
- Cai, L.; Ren, X.; Fu, X.; Peng, L.; Zeng, X. iEnhancer-XG: Interpretable sequence-based enhancers and their strength predictor. Bioinformatics 2021, 37, 1060–1067. [Google Scholar] [CrossRef]
- Khanal, J.; Tayara, H.; Chong, K.T. Identifying Enhancers and Their Strength by the Integration of Word Embedding and Convolution Neural Network. IEEE Access 2020, 8, 58369–58376. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Nguyen-Vo, T.H.; Le, N.Q.K.; Do, T.T.; Rahardja, S.; Nguyen, B.P. iEnhancer-ECNN: Identifying enhancers and their strength using ensembles of convolutional neural networks. BMC Genom. 2019, 20, 951. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Harwani, M.; Grama, A.; Chaterji, S. EP-DNN: A Deep Neural Network-Based Global Enhancer Prediction Algorithm. Sci. Rep. 2016, 6, 38433. [Google Scholar] [CrossRef]
- Kamran, H.; Tahir, M.; Tayara, H.; Chong, K.T. iEnhancer-Deep: A Computational Predictor for Enhancer Sites and Their Strength Using Deep Learning. Appl. Sci. 2022, 12, 2120. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Y.; Wan, S. Exploring Deep Learning for View-Based 3D Model Retrieval. ACM Trans. Multimed. Comput. Commun. Appl. (TOMM) 2020, 16, 1–21. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Song, R.; Sun, F. Nonlinear dictionary learning based deep neural networks. In Proceedings of the 2016 International Joint Conference on Neural Networks (IJCNN), Vancouver, BC, Canada, 24–29 July July 2016; pp. 3771–3776. [Google Scholar] [CrossRef]
- Geng, Q.; Yang, R.; Zhang, L. A deep learning framework for enhancer prediction using word embedding and sequence generation. Biophys. Chem. 2022, 286, 106822. [Google Scholar] [CrossRef]
- Niu, X.; Yang, K.; Zhang, G.; Yang, Z.; Hu, X. A Pretraining-Retraining Strategy of Deep Learning Improves Cell-Specific Enhancer Predictions. Front. Genet. 2020, 10, 1305. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Chen, N.; Chen, T.; Jiang, R. DeepEnhancer: Predicting enhancers by convolutional neural networks. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 637–644. [Google Scholar] [CrossRef]
- Rubin, C.J.; Megens, H.J.; Barrio, Á.M.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, Ö.; Jern, P.; Jørgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002.e13. [Google Scholar] [CrossRef]
- Längin, M.; Mayr, T.; Reichart, B.; Michel, S.; Buchholz, S.; Guethoff, S.; Dashkevich, A.; Baehr, A.; Egerer, S.; Bauer, A.; et al. Consistent success in life-supporting porcine cardiac xenotransplantation. Nature 2018, 564, 430–433. [Google Scholar] [CrossRef]
- Ekser, B.; Li, P.; Cooper, D.K.C. Xenotransplantation: Past, present, and future. Curr. Opin. Organ Tran. 2017, 22, 513–521. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, T.; Zhu, Y.; Ling, Z.; Yang, B.; Huang, L. A comparative investigation on H3K27ac enhancer activities in the brain and liver tissues between wild boars and domesticated pigs. Evol. Appl. 2022, 15, 1281–1290. [Google Scholar] [CrossRef]
- Zhao, Y.; Hou, Y.; Xu, Y.; Luan, Y.; Zhou, H.; Qi, X.; Hu, M.; Wang, D.; Wang, Z.; Fu, Y.; et al. A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome. Nat. Commun. 2021, 12, 2217. [Google Scholar] [CrossRef]
- Pan, Z.; Yao, Y.; Yin, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, G.; Trakooljul, N.; et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021, 12, 5848. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2010, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef] [PubMed]
- Oubounyt, M.; Louadi, Z.; Tayara, H.; Chong, K.T. DeePromoter: Robust Promoter Predictor Using Deep Learning. Front. Genet. 2019, 10, 286. [Google Scholar] [CrossRef]
- Sethi, A.; Gu, M.; Gumusgoz, E.; Chan, L.; Yan, K.K.; Rozowsky, J.; Barozzi, I.; Afzal, V.; Akiyama, J.A.; Plajzer-Frick, I.; et al. Supervised enhancer prediction with epigenetic pattern recognition and targeted validation. Nat. Methods 2020, 17, 807–814. [Google Scholar] [CrossRef]
- Yang, B.; Liu, F.; Ren, C.; Ouyang, Z.; Xie, Z.; Bo, X.; Shu, W. BiRen: Predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 2017, 33, 1930–1936. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, Y.; Zhang, Z.; Jiang, T.; Ling, Z.; Yang, B.; Li, W. Comparative Analysis of Promoters and Enhancers in the Pituitary Glands of the Bama Xiang and Large White Pigs. Front. Genet. 2021, 12, 697994. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Kang, H.; Luo, J.; Zhang, Y. A Comparative Analysis of Super-Enhancers and Broad H3K4me3 Domains in Pig, Human, and Mouse Tissues. Front. Genet. 2021, 12, 701049. [Google Scholar] [CrossRef]
- Deng, D.; Tan, X.; Han, K.; Ren, R.; Cao, J.; Yu, M. Transcriptomic and ChIP-seq Integrative Analysis Reveals Important Roles of Epigenetically Regulated lncRNAs in Placental Development in Meishan Pigs. Genes 2020, 11, 397. [Google Scholar] [CrossRef]
- MacPhillamy, C.; Alinejad-Rokny, H.; Pitchford, W.S.; Low, W.Y. Cross-species enhancer prediction using machine learning. Genomics 2022, 114, 110454. [Google Scholar] [CrossRef] [PubMed]
- Prowse-Wilkins, C.P.; Wang, J.; Xiang, R.; Garner, J.B.; Goddard, M.E.; Chamberlain, A.J. Putative Causal Variants Are Enriched in Annotated Functional Regions From Six Bovine Tissues. Front. Genet. 2021, 12, 664379. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, S.; Liu, M.; Kang, X.; Lin, S.; Li, B.; Connor, E.E.; Baldwin, R.L.; Tenesa, A.; Ma, L.; et al. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol. 2019, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.; Lukk, M.; Pignatelli, M.; Park, T.; Deaville, R.; Erichsen, J.; Jasinska, A.; et al. Enhancer Evolution across 20 Mammalian Species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef]
- Zhou, H.; Pan, Z.; Yao, Y.; Ying, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, K.; et al. Pig genome functional annotation enhances biological interpretations of complex traits and comparative epigenomics. Nat. Commun. 2021. [Google Scholar] [CrossRef]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2019, 48, D58. [Google Scholar] [CrossRef]
- Warr, A.; Affara, N.; Aken, B.; Beiki, H.; Bickhart, D.M.; Billis, K.; Chow, W.; Eory, L.; Finlayson, H.A.; Flicek, P.; et al. An improved pig reference genome sequence to enable pig genetics and genomics research. GigaScience 2019, 9, giaa051. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Habibi, M.; Weber, L.; Neves, M.; Wiegandt, D.L.; Leser, U. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics 2017, 33, i37–i48. [Google Scholar] [CrossRef]
- Hamid, M.N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019, 35, 2009–2016. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Bembom, O. seqLogo: An R Package for Plotting DNA Sequence Logos; R Package: Vienna, Austria, 2007. [Google Scholar]
- Fushiki, T. Estimation of prediction error by using K-fold cross-validation. Stat. Comput. 2009, 21, 137–146. [Google Scholar] [CrossRef]
- Jia, C.; He, W. EnhancerPred: A predictor for discovering enhancers based on the combination and selection of multiple features. Sci. Rep. 2016, 6, 38741. [Google Scholar] [CrossRef]
- Liu, B.; Li, K.; Huang, D.; Chou, K. iEnhancer-EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef]
- Niu, K.; Luo, X.; Zhang, S.; Teng, Z.; Zhang, T.; Zhao, Y. iEnhancer-EBLSTM: Identifying Enhancers and Strengths by Ensembles of Bidirectional Long Short-Term Memory. Front. Genet. 2021, 12, 665498. [Google Scholar] [CrossRef]
- Hasan, M.K.; Alam, M.A.; Dahal, L.; Roy, S.; Wahid, S.R.; Elahi, M.T.E.; Martí, R.; Khanal, B. Challenges of deep learning methods for COVID-19 detection using public datasets. Informat. Med. Unlocked 2022, 30, 100945. [Google Scholar] [CrossRef] [PubMed]
- Crowther, P.S.; Cox, R.J. Accuracy of neural network classifiers as a property of the size of the data set. In Proceedings of the International Conference on Knowledge-Based and Intelligent Information and Engineering Systems, Bournemouth, UK, 9–11 October 2006; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1143–1149. [Google Scholar] [CrossRef]
- Fang, J. A critical review of five machine learning-based algorithms for predicting protein stability changes upon mutation. Brief. Bioinform. 2019, 21, 1285–1292. [Google Scholar] [CrossRef]
- Bu, H.; Gan, Y.; Wang, Y.; Zhou, S.; Guan, J. A new method for enhancer prediction based on deep belief network. BMC Bioinf. 2017, 18, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.G.; Phan, D.; Lumbanraja, F.R.; Faisal, M.R.; Abapihi, B.; Purnama, B.; Delimayanti, M.K.; Mahmudah, K.R.; Kubo, M.; Satou, K. Applying Deep Learning Models to Mouse Behavior Recognition. J. Biomed. Sci. Eng. 2019, 12, 183–196. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Higgins, G.A.; Reamaroon, N.; Soroushmehr, S.; Allyn-Feuer, A.; Dinov, I.D.; Najarian, K.; Athey, B.D. Deep learning in pharmacogenomics: From gene regulation to patient stratification. Pharmacogenomics 2018, 19, 629–650. [Google Scholar] [CrossRef] [PubMed]
- Taskiran, I.I.; Spanier, K.I.; Christiaens, V.; Mauduit, D.; Aerts, S. Cell type directed design of synthetic enhancers. bioRxiv 2022. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Patil, S.S.; Pumphrey, M.O.; Carter, A.H. Multi-Trait Machine and Deep Learning Models for Genomic Selection using Spectral Information in a Wheat Breeding Program. bioRxiv 2021. [Google Scholar] [CrossRef]
- Telenti, A.; Lippert, C.; Chang, P.C.; DePristo, M. Deep learning of genomic variation and regulatory network data. Hum. Mol. Genet. 2018, 27, R63–R71. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Liu, H.; Gao, Y.; Liu, Y.; Chen, L.; Liu, L.; Irwin, D.M.; Hou, C.; Zhou, Z.A. Genome-wide identification of functional enhancers and their potential roles in pig breeding. J. Anim. Sci. Biotechnol. 2022, 13, 75. [Google Scholar] [CrossRef]
- Zeng, R.; Liao, M. Developing a Multi-Layer Deep Learning Based Predictive Model to Identify DNA N4-Methylcytosine Modifications. Front. Bioeng. Biotechnol. 2020, 8, 274. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer (Type) | Output Shape | Param |
|---|---|---|
| Conv1d-1 | [−1, 32, 200] | 800 |
| BatchNorm1d-2 | [−1, 32, 200] | 64 |
| Conv1d-3 | [−1, 32, 200] | 3104 |
| BatchNorm1d-4 | [−1, 32, 200] | 64 |
| Conv1d-5 | [−1, 32, 200] | 3104 |
| BatchNorm1d-6 | [−1, 32, 200] | 64 |
| MaxPool1d-7 | [−1, 32, 50] | 0 |
| Conv1d-8 | [−1, 64, 50] | 6208 |
| BatchNorm1d-9 | [−1, 64, 50] | 128 |
| Conv1d-10 | [−1, 64, 50] | 12,352 |
| BatchNorm1d-11 | [−1, 64, 50] | 128 |
| Conv1d-12 | [−1, 64, 50] | 12,352 |
| BatchNorm1d-13 | [−1, 64, 50] | 128 |
| MaxPool1d-14 | [−1, 64, 12] | 0 |
| Linear-15 | [−1, 256] | 196,864 |
| Linear-16 | [−1, 1] | 257 |
| Model | Accuracy Score | AUC Score | Sensitivity | Specificity |
|---|---|---|---|---|
| Model 1 (Parts 2, 3, 4, 5: Part 1) | 0.909626719 | 0.939438503 | 0.963326785 | 0.855926654 |
| Model 2 (Parts 1, 3, 4, 5: Part 2) | 0.910936477 | 0.944208139 | 0.974459725 | 0.847413229 |
| Model 3 (Parts 1, 2, 4, 5: Part 3) | 0.910609037 | 0.94386183 | 0.965291421 | 0.855926654 |
| Model 4 (Parts 1, 2, 3, 5: Part 4) | 0.910936477 | 0.940875633 | 0.964636542 | 0.857236411 |
| Model 5 (Parts 1, 2, 3, 4: Part 5) | 0.904715128 | 0.94601431 | 0.948264571 | 0.861165684 |
| Ensemble Model | 0.916502947 | 0.948383796 | 0.974459725 | 0.858546169 |
| Method | ACC | AUC | SN | SP | Source |
|---|---|---|---|---|---|
| iEnhancer-2L | 0.730 | 0.806 | 0.710 | 0.750 | Liu et al., 2016 [30] |
| EnhancerPred | 0.740 | 0.801 | 0.735 | 0.745 | Jia and He, 2016 [76] |
| iEnhancer-EL | 0.748 | 0.817 | 0.710 | 0.785 | Liu et al., 2018 [77] |
| iEnhancer-EBLSTM | 0.772 | 0.835 | 0.755 | 0.795 | Niu et al., 2021 [78] |
| PorcineAI-enhancer | 0.652 | 0.811 | 0.335 | 0.969 | This study |
| PorcineAI-enhancer (human enhancer data) | 0.769 | 0.832 | 0.785 | 0.752 | This study |
| Tissue | Pig | Human |
|---|---|---|
| Heart | 0.8240 | 0.7031 |
| iPSC | 0.2606 | 0.3146 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhang, H.; Chen, N.; Zeng, T.; Ai, X.; Wu, K. PorcineAI-Enhancer: Prediction of Pig Enhancer Sequences Using Convolutional Neural Networks. Animals 2023, 13, 2935. https://doi.org/10.3390/ani13182935
Wang J, Zhang H, Chen N, Zeng T, Ai X, Wu K. PorcineAI-Enhancer: Prediction of Pig Enhancer Sequences Using Convolutional Neural Networks. Animals. 2023; 13(18):2935. https://doi.org/10.3390/ani13182935
Chicago/Turabian StyleWang, Ji, Han Zhang, Nanzhu Chen, Tong Zeng, Xiaohua Ai, and Keliang Wu. 2023. "PorcineAI-Enhancer: Prediction of Pig Enhancer Sequences Using Convolutional Neural Networks" Animals 13, no. 18: 2935. https://doi.org/10.3390/ani13182935