
Runs of Homozygosity Analysis Reveals Genomic Diversity and Population Structure of an Indigenous Cattle Breed in Southwest China
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sample Collection
2.3. Genomic DNA and Sequencing
2.4. SNP Genotyping and Quality Controls
2.5. Genetic Diversity and Runs of Homozygosity
2.6. Functional Analysis
3. Results
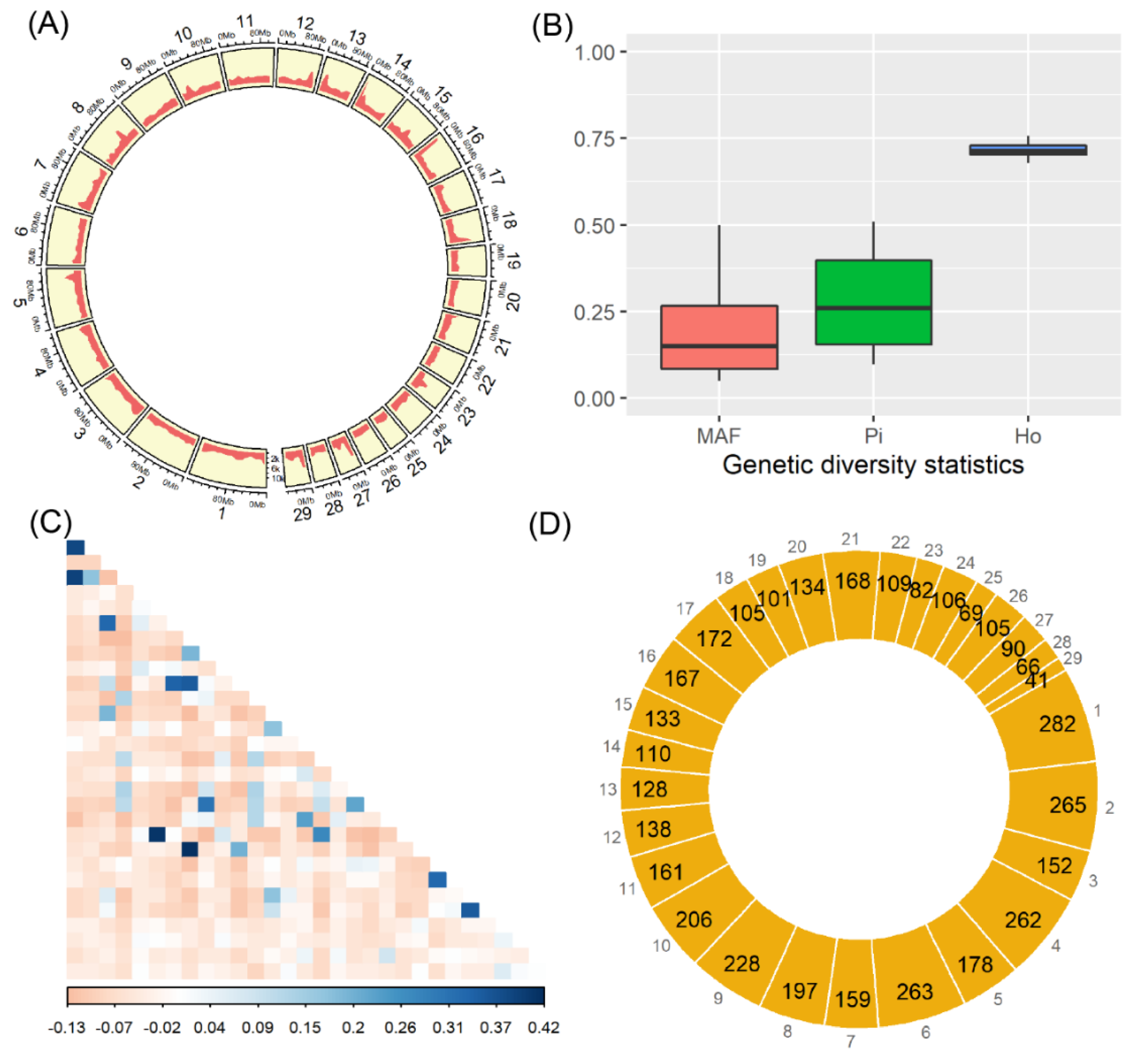
3.1. SNPs and Genetic Diversity
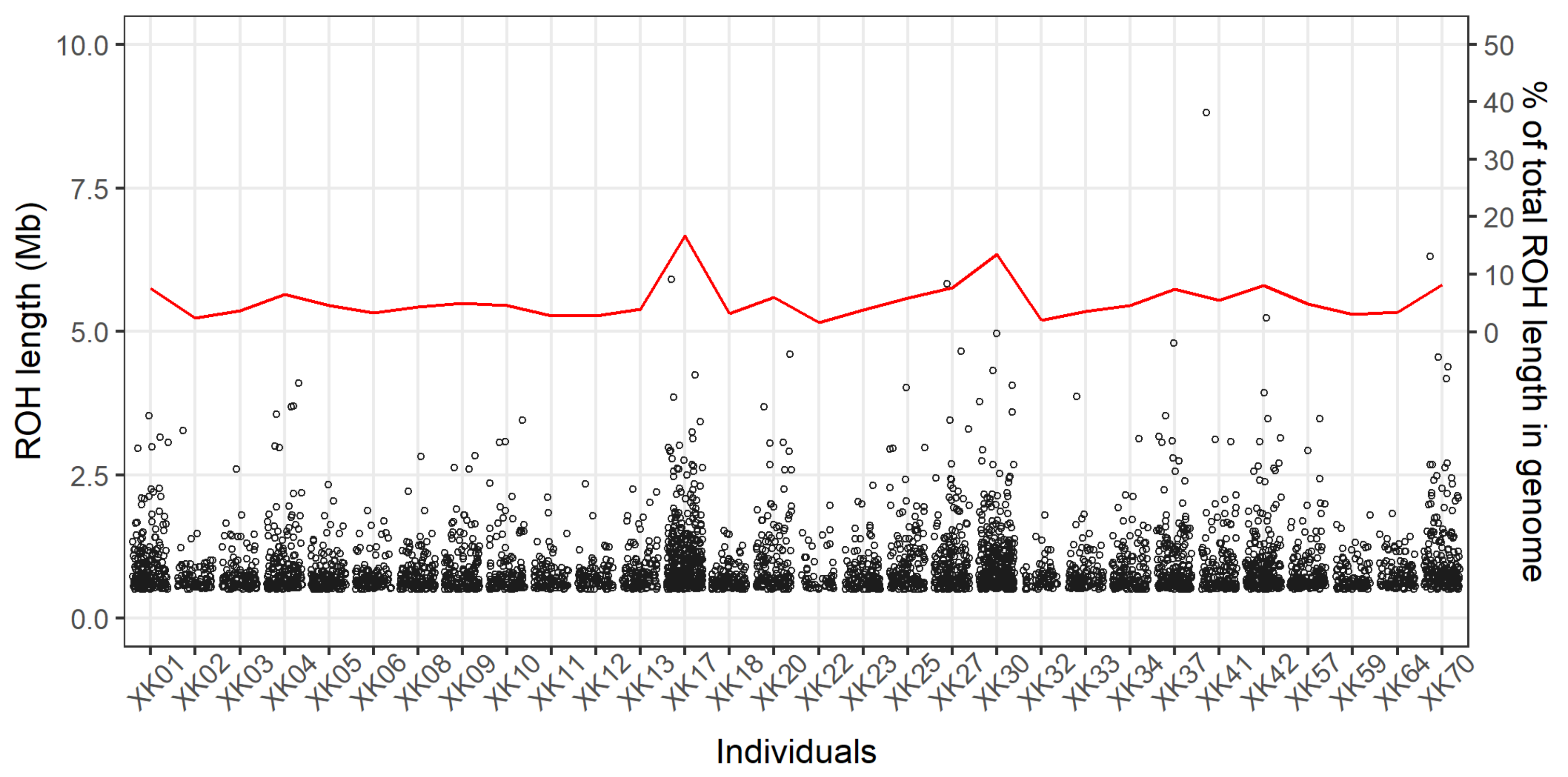
3.2. Genomic Patterns of Runs of Homozygosity
3.3. Genes within the Significant Genomic Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kristensen, T.N.; Hoffmann, A.A.; Pertoldi, C.; Stronen, A.V. What can livestock breeders learn from conservation genetics and vice versa? Front. Genet. 2015, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Gan, J.; Fang, D.; Tang, H.; Wang, H.; Yi, J.; Fu, M. Genome-wide SNP discovery and evaluation of genetic diversity among six Chinese indigenous cattle breeds in Sichuan. PLoS ONE 2018, 13, e0201534. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Weber, J.L. Long homozygous chromosomal segments in reference families from the centre d’Etude du polymorphisme humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.G. The size distribution of homozygous segments in the human genome. Am. J. Hum. Genet. 1999, 65, 1489–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Alemu, S.W.; Kadri, N.K.; Harland, C.; Faux, P.; Charlier, C.; Caballero, A.; Druet, T. An evaluation of inbreeding measures using a whole-genome sequenced cattle pedigree. Heredity 2021, 126, 410–423. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef] [Green Version]
- Nani, J.P.; Peñagaricano, F. Whole-genome homozygosity mapping reveals candidate regions affecting bull fertility in US Holstein cattle. BMC Genom. 2020, 21, 338. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Jasielczuk, I.; Ząbek, T.; Ropka-Molik, K.; Litwińczuk, Z.; Bugno-Poniewierska, M. A comprehensive analysis of runs of homozygosity of eleven cattle breeds representing different production types. Animals 2019, 9, 1024. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Liu, Y.; Niu, Q.; Zheng, X.; Zhang, T.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; et al. Runs of homozygosity analysis reveals consensus homozygous regions affecting production traits in Chinese Simmental beef cattle. BMC Genom. 2021, 22, 678. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. 2019. R Package Version 0.9.6. Available online: https://CRAN.R-project.org/package=detectRUNS (accessed on 20 May 2022).
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, G.; Wan, R.; Wang, X.; Wang, J.; Li, P. Atmospheric Thermal and Dynamic Vertical Structures of Summer Hourly Precipitation in Jiulong of the Tibetan Plateau. Atmosphere 2021, 12, 505. [Google Scholar] [CrossRef]
- Calboli, F.C.; Sampson, J.; Fretwell, N.; Balding, D.J. Population structure and inbreeding from pedigree analysis of purebred dogs. Genetics 2008, 179, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A publicly available repository of ROH islands reveals signatures of selection in different livestock and pet species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef]
- Cheruiyot, E.K.; Haile-Mariam, M.; Cocks, B.G.; MacLeod, I.M.; Xiang, R.; Pryce, J.E. New loci and neuronal pathways for resilience to heat stress in cattle. Sci. Rep. 2021, 11, 16619. [Google Scholar] [CrossRef]
- Marín-Garzón, N.A.; Magalhães, A.F.B.; Mota, L.F.M.; Fonseca, L.F.S.; Chardulo, L.A.L.; Albuquerque, L.G. Genome-wide association study identified genomic regions and putative candidate genes affecting meat color traits in Nellore cattle. Meat Sci. 2021, 171, 108288. [Google Scholar] [CrossRef]
- Gholap, P.N.; Kale, D.S.; Sirothia, A.R. Genetic diseases in cattle: A review. Res. J. Anim. Vet. Fish. Sci. 2014, 2, 24–33. [Google Scholar]
- Nordmark, G.; Kristjansdottir, G.; Theander, E.; Appel, S.; Eriksson, P.; Vasaitis, L.; Kvarnström, M.; Delaleu, N.; Lundmark, P.; Lundmark, A.; et al. Association of EBF1, FAM167A (C8orf13)-BLK and TNFSF4 gene variants with primary Sjögren’s syndrome. Genes Immun. 2011, 12, 100–109. [Google Scholar] [CrossRef]
- Bhati, M.; Kadri, N.K.; Crysnanto, D.; Pausch, H. Assessing genomic diversity and signatures of selection in Original Braunvieh cattle using whole-genome sequencing data. BMC Genom. 2020, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, J.W.; Reecy, J.M.; Garrick, D.J.; Duan, Q.; Beitz, D.C.; Koltes, J.E.; Saatchi, M.; Koesterke, L.; Mateescu, R.G. Deriving gene networks from SNP associated with triacylglycerol and phospholipid fatty acid fractions from Ribeyes of Angus cattle. Front. Genet. 2016, 7, 116. [Google Scholar] [CrossRef] [Green Version]
- Yoon, D.; Ko, E. Association study between SNPs of the genes within bovine QTLs and meat quality of Hanwoo. J. Anim. Sci. 2016, 94, 145. [Google Scholar] [CrossRef] [Green Version]
- Mei, C.G.; Gui, L.S.; Wang, H.C.; Tian, W.Q.; Li, Y.K.; Zan, L.S. Polymorphisms in adrenergic receptor genes in Qinchuan cattle show associations with selected carcass traits. Meat Sci. 2018, 135, 166–173. [Google Scholar] [CrossRef]
- Costilla, R.; Kemper, K.E.; Byrne, E.M.; Porto-Neto, L.R.; Carvalheiro, R.; Purfield, D.C.; Doyle, J.L.; Berry, D.P.; Moore, S.S.; Wray, N.R.; et al. Genetic control of temperament traits across species: Association of autism spectrum disorder risk genes with cattle temperament. Genet. Sel. Evol. 2020, 52, 51. [Google Scholar] [CrossRef]
- Manca, E.; Cesarani, A.; Falchi, L.; Atzori, A.S.; Gaspa, G.; Rossoni, A.; Macciotta, N.P.P.; Dimauro, C. Genome-wide association study for residual concentrate intake using different approaches in Italian Brown Swiss. Ital. J. Anim. Sci. 2021, 20, 1957–1967. [Google Scholar] [CrossRef]
- Sanglard, L.P.; Nascimento, M.; Moriel, P.; Sommer, J.; Ashwell, M.; Poore, M.H.; Duarte, M.D.S.; Serão, N.V. Impact of energy restriction during late gestation on the muscle and blood transcriptome of beef calves after preconditioning. BMC Genom. 2018, 19, 702. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, L.; Gu, Y.; Chang, Y.; Liang, C.; Guo, X.; Bao, P.; Chu, M.; Ding, X.; Yan, P. Bta-miR-2400 Targets SUMO1 to Affect Yak Preadipocytes Proliferation and Differentiation. Biology 2021, 10, 949. [Google Scholar] [CrossRef]
- Zhang, W.W.; Tong, H.L.; Sun, X.F.; Hu, Q.; Yang, Y.; Li, S.F.; Yan, Y.Q.; Li, G.P. Identification of miR-2400 gene as a novel regulator in skeletal muscle satellite cells proliferation by targeting MYOG gene. Biochem. Biophys. Res. Commun. 2015, 463, 624–631. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genomic Regions | N of SNPs | Candidate Genes | |||
|---|---|---|---|---|---|
| Chr | Start | End | Kb | ||
| BTA2 | 81,899,298 | 82,248,318 | 349 | 65 | None |
| 82,355,133 | 82,378,221 | 23 | 7 | None | |
| BTA6 | 66,234,825 | 66,436,164 | 201 | 57 | ATP10D, CORIN |
| 81,161,826 | 81,281,680 | 120 | 22 | EPHA5 | |
| BTA9 | 46,061,002 | 46,211,676 | 151 | 38 | None |
| 46,270,931 | 46,319,445 | 49 | 8 | None | |
| BTA11 | 14,444,882 | 14,944,596 | 500 | 89 | MEMO1, DPY30, SPAST, SLC30A6, NLRC4, YIPF4 |
| BTA15 | 11,474,883 | 12,369,497 | 895 | 152 | None |
| BTA16 | 40,264,305 | 40,823,586 | 559 | 92 | TNFSF18, TNFSF4 |
| 43,872,679 | 44,009,452 | 137 | 18 | TMEM201, SLC25A33 | |
| BTA17 | 15,394,915 | 15,766,365 | 371 | 64 | INPP4B |
| BTA21 | 2,945,936 | 3,556,313 | 610 | 136 | None |
| 4,051,182 | 4,754,672 | 703 | 129 | SCAPER, RCN2, PSTPIP1, TSPAN3, PEAK1 | |
| 31,947,095 | 32,283,141 | 336 | 70 | PEAK1 | |
| 32,283,208 | 32,346,053 | 63 | 15 | PEAK1, HMG20A, LINGO1 | |
| 32,490,146 | 32,802,506 | 312 | 89 | None | |
| 36,931,645 | 37,537,467 | 606 | 150 | GABRB3, GABRA5 | |
| BTA27 | 32,891,817 | 33,308,815 | 417 | 56 | ZNF703, ERLIN2, PLPBP, ADGRA2, BRF2, RAB11FIP1, GOT1L1, ADRB3, EIF4EBP1, ASH2L |
| 33,448,422 | 33,466,272 | 18 | 6 | NSD3, bta-mir-2400 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Shi, Y.; He, F.; Fang, D.; Gan, J.; Wu, F.; AG, Y.; Deng, X.; Cao, Q.; Duo, C.; et al. Runs of Homozygosity Analysis Reveals Genomic Diversity and Population Structure of an Indigenous Cattle Breed in Southwest China. Animals 2022, 12, 3239. https://doi.org/10.3390/ani12233239
Wang W, Shi Y, He F, Fang D, Gan J, Wu F, AG Y, Deng X, Cao Q, Duo C, et al. Runs of Homozygosity Analysis Reveals Genomic Diversity and Population Structure of an Indigenous Cattle Breed in Southwest China. Animals. 2022; 12(23):3239. https://doi.org/10.3390/ani12233239
Chicago/Turabian StyleWang, Wei, Yi Shi, Fang He, Donghui Fang, Jia Gan, Fuqiu Wu, Yueda AG, Xiaodong Deng, Qi Cao, Chu Duo, and et al. 2022. "Runs of Homozygosity Analysis Reveals Genomic Diversity and Population Structure of an Indigenous Cattle Breed in Southwest China" Animals 12, no. 23: 3239. https://doi.org/10.3390/ani12233239