
The Relationship between Brachionus calyciflorus-Associated Bacterial and Bacterioplankton Communities in a Subtropical Freshwater Lake
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Treatment
2.2. Sequence Determination of the 16S Gene from the Bacterial Community
2.3. Data Analysis
3. Results
3.1. Alpha and Beta Diversity of AB and NB Communities
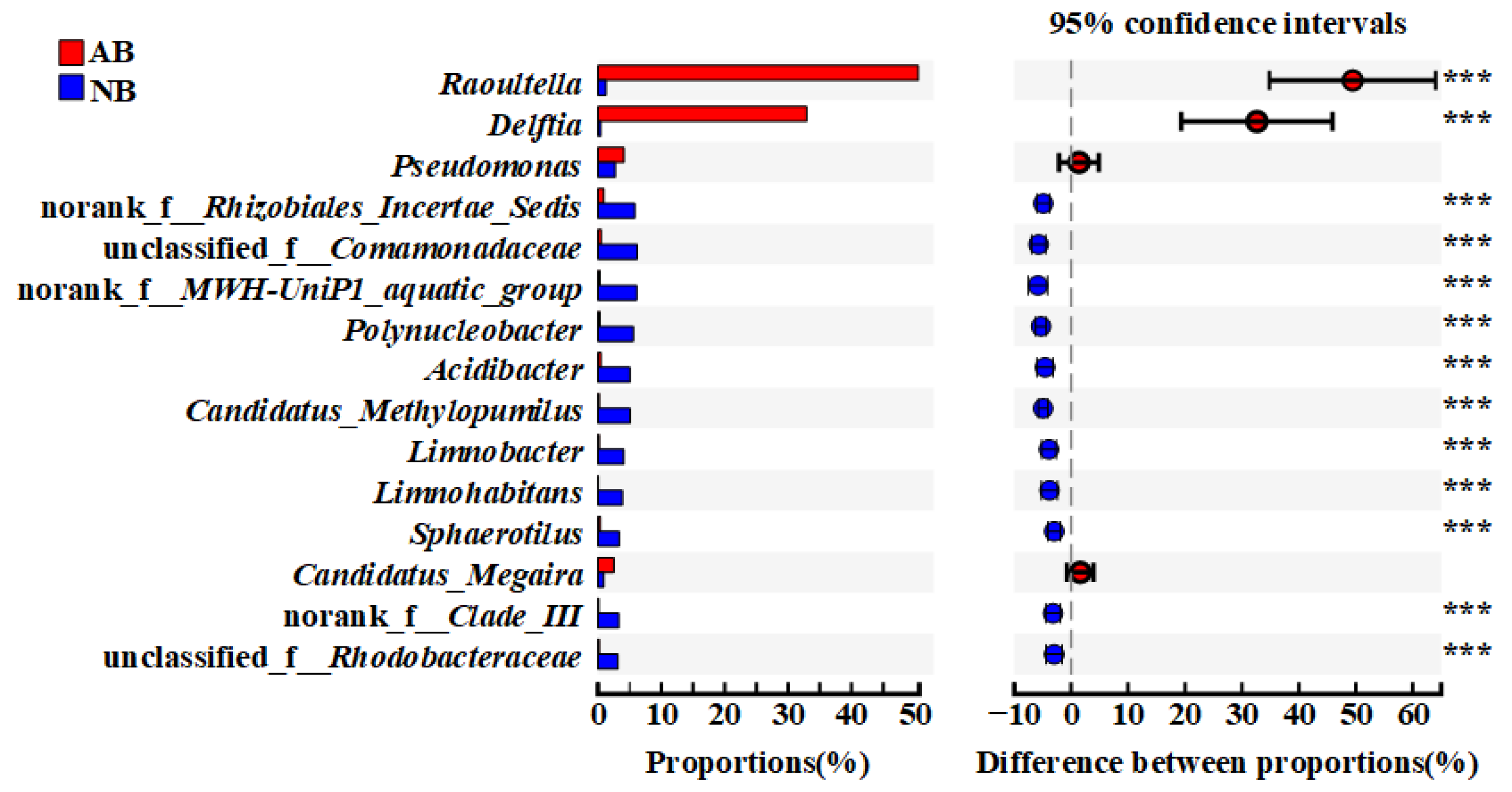
3.2. Differences in Community Composition of the AB and NB Groups
3.3. Significantly Different OTUs between AB and NB Communities
3.4. Relationship between Bacterial Community and Environmental Parameters
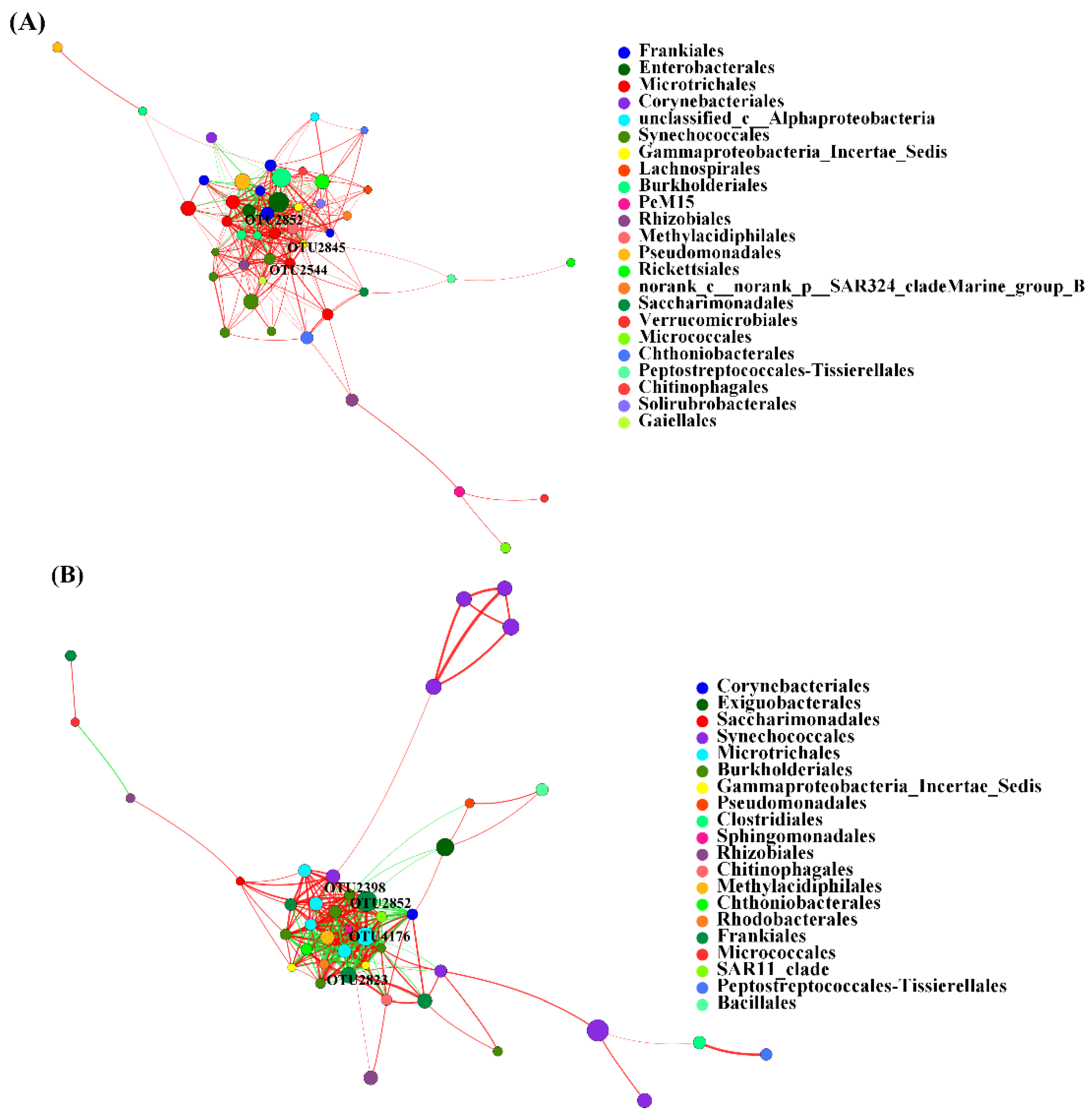
3.5. Co-Occurrence Network of AB and NB Communities
3.6. Functional Characteristics of AB and NB Communities
4. Discussion
4.1. Community Composition Difference and Keystone Taxa between AB and NB Communities
4.2. Environmental Regulation of AB and NB Communities
4.3. Main Functions of the B. Calyciflorus-Associated Bacterial Community
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Špoljar, M.; Dražina, T.; Lajtner, J.; Sertić, M.D.; Radanović, I.; Wallace, R.L.; Matulic, D.; Tomlijanovic, T. Zooplankton assemblage in four temperate shallow waterbodies in association with habitat heterogeneity and alternative states. Limnologica 2018, 71, 51–61. [Google Scholar] [CrossRef]
- Nam, Y.D.; Sung, Y.; Chang, H.W.; Roh, S.W.; Kim, K.H.; Rhee, S.K.; Kim, J.-C.; Kim, J.-Y.; Yoon, J.-K.; Bae, J.-W. Characterization of the depth-related changes in the microbial communities in Lake Hovsgol sediment by 16S rRNA gene-based approaches. J. Microbiol. 2008, 46, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltar, F.; Stuck, E.; Morales, S.; Currie, K. Bacterioplankton carbon cycling along the subtropical frontal zone off New Zealand. Prog. Oceanogr. 2015, 135, 168–175. [Google Scholar] [CrossRef]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Turk, V.; Grossart, H.P. Linkage between crustacean zooplankton and aquatic bacteria. Aquat. Microb. Ecol. 2010, 61, 261–277. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hao, Z.; Ding, R.; Li, H.; Tang, X.; Chen, F. Host Dependence of Zooplankton-Associated Microbes and Their Ecological Implications in Freshwater Lakes. Water 2021, 13, 2949. [Google Scholar] [CrossRef]
- Hansen, B.; Bech, G. Bacteria associated with a marine planktonic copepod in culture. I. bacterial genera in seawater, body surface, intestines and fecal pellets and succession during fecal pellet degradation. J. Plankton Res. 1996, 18, 257–273. [Google Scholar] [CrossRef]
- Grossart, H.P.; Dziallas, C.; Tang, K.W. Bacterial diversity associated with freshwater zooplankton. Environ. Microbiol. Rep. 2009, 1, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Corte, D.D.; Srivastava, A.; Koski, M.; Gracia, J.A.L.; Takaki, Y.; Yokokawa, T.; Nunoura, T.; Elizabeth, N.H.; Sintes, E.; Herndl, G.J. MetaSgenomic insights into zooplankton-associated bacterial communities. Environ. Microbiol. 2018, 20, 492–505. [Google Scholar] [CrossRef]
- Møller, E.F.; Riemann, L.; Søndergaard, M. Bacteria associated with copepods: Abundance, activity and community composition. Aquat. Microb. Ecol. 2007, 47, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Corte, D.D.; Lekunberri, I.; Sintes, E.; Garcia, J.A.L.; Gonzales, S.; Herndl, G.J. Linkage between copepods and bacteria in the North Atlantic Ocean. Aquat. Microb. Ecol. 2014, 72, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Grossart, H.P.; Dziallas, C.; Leunert, F.; Tang, K.W. Bacteria dispersal by hitchhiking on zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 11959–11964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Chen, W.; Sun, J.; Liu, L.; Huang, X. Spatial variation in bacterioplankton communities in the Pearl River, South China: Impacts of land use and physicochemical factors. Microorganisms 2020, 8, 814. [Google Scholar] [CrossRef]
- Callens, M.; Watanabe, H.; Kato, Y.; Miura, J.; Decaestecker, E. Microbiota inoculum composition affects holobiont assembly and host growth in Daphnia. Microbiome 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Dattagupta, S.; Schaperdoth, I.; Montanari, A.; Mariani, S.; Kita, N.; Valley, J.W.; Macalady, J.L. A novel symbiosis between chemoautotrophic bacteria and a freshwater cave amphipod. ISME J. 2009, 3, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, K.M.; Moisander, P.H. Microbial diversity associated with copepods in the North Atlantic subtropical gyre. FEMS Microbiol. Ecol. 2015, 91, fiv064. [Google Scholar] [CrossRef] [Green Version]
- Akbar, S.; Huang, J.; Zhou, Q.; Gu, L.; Sun, Y.; Zhang, L.; Lyu, K.; Yang, Z. Elevated temperature and toxic Microcystis reduce Daphnia fitness and modulate gut microbiota. Environ. Pollut. 2021, 271, 116409. [Google Scholar] [CrossRef]
- Herzig, A. The analysis of planktonic rotifer populations: A plea for long-term investigations. Rotifer Symp. IV 1987, 42, 163–180. [Google Scholar] [CrossRef]
- Zink, I.C.; Douillet, P.A.; Benetti, D.D. Improvement of rotifer Brachionus plicatilis population growth dynamics with inclusion of Bacillus spp. Probiotics. Aquacult. Res. 2013, 44, 200–211. [Google Scholar] [CrossRef]
- Turgay, E.; Steinum, T.M.; Eryalçın, K.M.; Yardımcı, R.E.; Karataş, S. The influence of diet on the microbiota of live-feed rotifers (Brachionus plicatilis) used in commercial fish larviculture. FEMS Microbiol. Lett. 2020, 367, 20. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.C.; Verschoor, A.M. Effects of food quality on life history of the rotifer Brachionus calyciflorus Pallas. Freshwater Biol. 2004, 49, 1138–1151. [Google Scholar] [CrossRef]
- Nandini, S.; Sánchez-Zamora, C.; Sarma, S.S.S. Toxicity of cyanobacterial blooms from the reservoir Valle de Bravo (Mexico): A case study on the rotifer Brachionus calyciflorus. Sci. Total Environ. 2019, 688, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Snell, T.W.; Yang, J. Ecological strategy of rotifer (Brachionus calyciflorus) exposed to predator- and competitor-conditioned media. Hydrobiologia 2011, 658, 163–171. [Google Scholar] [CrossRef]
- Michaloudi, E.; Papakostas, S.; Stamou, G.; Neděla, V.; Tihlaříková, E.; Zhang, W.; Declerck, S.A.J. Reverse taxonomy applied to the Brachionus calyciflorus cryptic species complex: Morphometric analysis confirms species delimitations revealed by molecular phylogenetic analysis and allows the (re)description of four species. PLoS ONE 2018, 13, e0203168. [Google Scholar] [CrossRef]
- SEPA. State EPA of China, Monitoring and Analysis Methods for Water and Wastewater, 4th ed.; China Environmental Science Press: Beijing, China, 2002. [Google Scholar]
- Field, J.G.; Clarke, K.R.; Warwick, R.M. A practical strategy for analysing multispecies distribution patterns. Mar. Ecol. Prog. Ser. 1982, 8, 37–52. [Google Scholar] [CrossRef]
- Thompson, C.G.; Kim, R.S.; Aloe, A.M.; Becker, B.J. Extracting the variance inflation factor and other multicollinearity diagnostics from typical regression results. Basic Appl. Soc. Psychol. 2017, 39, 81–90. [Google Scholar] [CrossRef]
- Adair, K.L.; Douglas, A.E. Making a microbiome: The many determinants of host-associated microbial community composition. Curr. Opin. Microbiol. 2017, 35, 23–29. [Google Scholar] [CrossRef]
- Wilkes Walburn, J.; Wemheuer, B.; Thomas, T.; Copeland, E.; O’Connor, W.; Booth, M.; Fielder, S.; Egan, S. Diet and diet-associated bacteria shape early microbiome development in Yellowtail Kingfish (Seriola lalandi). Microb. Biotechnol. 2019, 12, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.W. Copepods as microbial hotspots in the ocean: Effects of host feeding activities on attached bacteria. Aquat. Microb. Ecol. 2005, 38, 31–40. [Google Scholar] [CrossRef]
- Appel, T.M.; Quijano-Martínez, N.; Cadena, E.D.L.; Mojica, M.F.; Villegas, M.V. Microbiological and Clinical Aspects of Raoultella spp. Front. Public Health 2021, 9, 686789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xie, X.; Liu, Y.; Zheng, X.; Wang, Y.; Cong, J.; Yu, C.; Liu, N.; He, Z.; Liu, J.; et al. Sugar sources as Co-substrates promoting the degradation of refractory dye: A comparative study. Ecotoxicol. Environ. Saf. 2019, 184, 109613. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.X.; Yu, J.; Xiang, L.; Yu, L.Y.; Zhao, H.M.; Mo, C.H.; Li, Y.-W.; Cai, Q.-Y.; Wong, M.-H.; Li, Q.X. Co-metabolic degradation of the antibiotic ciprofloxacin by the enriched bacterial consortium XG and its bacterial community composition. Sci. Total Environ. 2019, 665, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Schicklberger, M.; Shapiro, N.; Loqué, D.; Woyke, T.; Chakraborty, R. Draft genome sequence of Raoultella terrigena R1Gly, a diazotrophic endophyte. Genome Announce. 2015, 3, e00607–e00615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, N.O.G.; Brandt, K.K.; Nybroe, O.; Hansen, M. Delftia lacustris sp. nov.; a peptidoglycan-degrading bacterium from fresh water, and emended description of Delftia tsuruhatensis as a peptidoglycan-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2009, 59, 2195–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Yun, S.H.; Lee, H.; Seo, G.; Kim, S.I. Multiomics analysis of aniline-degrading bacterium, Delftia sp. K82. J. Anal. Sci. Technol. 2021, 12, 6. [Google Scholar] [CrossRef]
- Urata, M.; Uchida, E.; Nojiri, H.; Omori, T.; Obo, R.; Miyaura, N.; Ouchiyama, N. Genes involved in aniline degradation by Delftia acidovorans strain 7N and its distribution in the natural environment. Biosci. Biotechnol. Biochem. 2004, 68, 2457–2465. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, C.S.; Sood, P.; Kanwar, S.S.; Sharma, P.N.; Kumar, A.; Mehta, P.K. Isolation and characterization of gut bacteria of fruit fly, Bactrocera tau (Walker). Phytoparasitica 2013, 41, 193–201. [Google Scholar] [CrossRef]
- Wu, J.Y.; Yan, M.C.; Sang, Y.; Li, F.; Luo, K.; Hu, L.H. Correlation between intestinal macrobiota and growth of white shrimp (Litopenaeus vannamei). Appl. Ecol. Environ. Res. 2021, 19, 4993–5005. [Google Scholar] [CrossRef]
- Kang, Y.J.; Diao, X.N.; Zhao, G.Y.; Chen, M.H.; Xiong, Y.; Shi, M.; Fu, W.M.; Guo, Y.-J.; Pan, B.; Chen, X.-P.; et al. Extensive diversity of Rickettsiales bacteria in two species of ticks from China and the evolution of the Rickettsiales. BMC Evol. Biol. 2014, 14, 167. [Google Scholar] [CrossRef]
- Lanzoni, O.; Plotnikov, A.; Khlopko, Y.; Munz, G.; Petroni, G.; Potekhin, A. The core microbiome of sessile ciliate Stentor coeruleus is not shaped by the environment. Sci. Rep. 2019, 9, 11356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzoni, O.; Sabaneyeva, E.; Modeo, L.; Castelli, M.; Lebedeva, N.; Verni, F.; Schrallhammer, M.; Potekhin, A.; Petroni, G. Diversity and environmental distribution of the cosmopolitan endosymbiont “Candidatus megaira”. Sci. Rep. 2019, 9, 1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, F.E.; Grosser, K.; Schrallhammer, M. Natural shifts in endosymbionts’ occurrence and relative frequency in their ciliate host population. Front. Microbiol. 2021, 12, 791615. [Google Scholar] [CrossRef] [PubMed]
- Tiralerdpanich, P.; Nasaree, S.; Pinyakong, O.; Sonthiphand, P. Variation of the mangrove sediment microbiomes and their phenanthrene biodegradation rates during the dry and wet seasons. Environ. Pollut. 2021, 289, 117849. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.J.A.; Bacosa, H.P.; Chien, M.F.; Inoue, C. Enhanced degradation of polycyclic aromatic hydrocarbons (PAHs) in the rhizosphere of sudangrass (Sorghum×drummondii). Chemosphere 2019, 234, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.W.; Liu, J.F.; Gu, J.D.; Mu, B.Z. Nitrate-reducing community in production water of three oil reservoirs and their responses to different carbon sources revealed by nitrate-reductase encoding gene (napA). Int. Biodeterior. Biodegrad. 2011, 65, 1081–1086. [Google Scholar] [CrossRef]
- Erlacher, A.; Cernava, T.; Cardinale, M.; Soh, J.; Sensen, C.W.; Grube, M.; Berg, G. Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front. Microbiol. 2015, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Hassen, A.I.; Swanevelder, Z.H.; Bopape, F.L.; Lamprecht, S.C. Draft genome sequence of Mesorhizobium sp. Strain SARCC-RB16n, an effective nodulating and nitrogen-fixing symbiont of rooibos Aspalathus linearis (Burm. f.). in South Africa. Microbiol. Resour. Announce. 2020, 9, e01187-19. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Bociu, I.; Kolton, M.; Huettel, M.; Kostla, J.E. Succession of microbial populations and nitrogen-fixation associated with the biodegradation of sediment-oil-agglomerates buried in a Florida sandy beach. Sci. Rep. 2019, 9, 19041. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.E.; Birrer, S.C.; Dafforn, K.A.; Mitrovic, S.M.; Crane, S.L.; Johnston, E.L.; Wemheuer, F.; Navarro, A.; Harrison, A.J.; Turner, I.L.; et al. Wastewater effluents cause microbial community shifts and change trophic status. Water Res. 2021, 200, 117206. [Google Scholar] [CrossRef]
- Soares, M.C.S.; Lürling, M.; Huszar, V.L.M. Responses of the rotifer Brachionus calyciflorus to two tropical toxic cyanobacteria (Cylindrospermopsis raciborskii and Microcystis aeruginosa) in pure and mixed diets with green algae. J. Plankton Res. 2010, 32, 999–1008. [Google Scholar] [CrossRef]
- Qin, Y.; Hou, J.; Deng, M.; Liu, Q.; Wu, C.; Ji, Y.; He, X. Bacterial abundance and diversity in pond water supplied with different feeds. Sci. Rep. 2016, 6, 35232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Xia, P.; Song, X.; Lin, T.; Cao, H. Community structure and functional diversity of epiphytic bacteria and planktonic bacteria on submerged macrophytes in Caohai Lake, southwest of China. Ann. Microbiol. 2019, 69, 933–944. [Google Scholar] [CrossRef]
- Elsheshtawy, A.; Clokie, B.G.J.; Albalat, A.; Beveridge, A.; Hamza, A.; Ibrahim, A.; MacKenzie, S. Characterization of external mucosal microbiomes of nile tilapia and grey mullet co-cultured in semi-intensive pond systems. Front. Microbiol. 2021, 12, 773860. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhou, A.; Wei, T.; Li, S.; Yang, B.; Xu, G.; Zou, J. Nanoplastics induce more serious microbiota dysbiosis and inflammation in the gut of adult zebrafish than microplastics. Bull. Environ. Contam. Toxicol. 2021, 107, 640–650. [Google Scholar] [CrossRef]
- Do, T.H.; Dao, T.K.; Nguyen, K.H.V.; Le, N.G.; Nguyen, T.M.P.; Le, T.L.; Phung, T.N.; Van Straalen, N.M.; Roelofs, D.; Truong, N.H. Metagenomic analysis of bacterial community structure and diversity of lignocellulolytic bacteria in Vietnamese native goat rumen. Asian-Australas. J. Anim. Sci. 2018, 31, 738–747. [Google Scholar] [CrossRef]
- Zou, Y.; Liang, N.; Zhang, X.; Han, C.; Nan, X. Functional differentiation related to decomposing complex carbohydrates of intestinal microbes between two wild zokor species based on 16SrRNA sequences. BMC Vet. Res. 2021, 17, 216. [Google Scholar] [CrossRef]
- Qadir, R.M.; Assafi, M.S. The association between body mass index and the oral Firmicutes and Bacteroidetes profiles of healthy individuals. Malays. Fam. Physician 2021, 16, 36–43. [Google Scholar] [CrossRef]
- Filippo, D.C.; Cavalieri, D.; Paola, D.M.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Ramayo-Caldas, Y.; Mach, N.; Lepage, P.; Levenez, F.; Denis, C.; Lemonnier, G.; Leplat, J.-J.; Billon, Y.; Berri, M.; Dore, J.; et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 2016, 10, 2973–2977. [Google Scholar] [CrossRef]
- Si, J.; You, H.J.; Yu, J.; Sung, J.; Ko, G. Prevotella as a hub for vaginal microbiota under the influence of host genetics and their association with obesity. Cell Host Microbe 2017, 21, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shi, R.; Han, R.; Ji, M.; Xu, X.; Wang, G. Community structure of epiphytic bacteria on Potamogeton pectinatus and the surrounding bacterioplankton in Hongze Lake. Mar. Freshwater Res. 2021, 72, 997–1003. [Google Scholar] [CrossRef]
- Eckert, E.M.; Amalfitano, S.; Cesare, A.D.; Manzari, C.; Corno, G.; Fontaneto, D. Different substrates within a lake harbour connected but specialised microbial communities. Hydrobiologia 2020, 847, 1689–1704. [Google Scholar] [CrossRef]
- Eckert, E.M.; Anicic, N.; Fontaneto, D. Freshwater zooplankton microbiome composition is highly flexible and strongly influenced by the environment. Mol. Ecol. 2021, 30, 1545–1558. [Google Scholar] [CrossRef]
- Ma, Z.; Wen, X.; Zhao, F.; Xia, Y.; Huang, X.; Waite, D.; Guan, J. Effect of temperature variation on membrane fouling and microbial community structure in membrane bioreactor. Bioresour. Technol. 2013, 133, 462–468. [Google Scholar] [CrossRef]
- Xia, X.; Stewart, D.I.; Cheng, L.; Wang, K.; Li, J.; Zhang, D.; Ding, A. Changes in groundwater bacterial community during cyclic groundwater-table variations. Hydrol. Processes 2020, 34, 4973–4984. [Google Scholar] [CrossRef]
- Wietz, M.; Wemheuer, B.; Simon, H.; Giebel, H.A.; Seibt, M.A.; Daniel, R.; Brinkhoff, T.; Simon, M. Bacterial community dynamics during polysaccharide degradation at contrasting sites in the Southern and Atlantic Oceans. Environ. Microbiol. 2015, 17, 3822–3831. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Wu, Y.; Gao, T.; Liu, C. Community structure and function of epiphytic bacteria associated with myriophyllum spicatum in Baiyangdian Lake, China. Front Microbiol. 2021, 12, 705509. [Google Scholar] [CrossRef]
- Yuan, J.; Ma, J.; Sun, Y.; Zhou, T.; Zhao, Y.; Yu, F. Microbial degradation and other environmental aspects of microplastics/plastics. Sci. Total Environ. 2020, 715, 136968. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, I. Role of microbiome and biofilm in environmental plastic degradation. Biocatal. Agric. Biotechnol. 2022, 39, 102263. [Google Scholar] [CrossRef]
- Zhu, F.; Doyle, E.; Zhu, C.; Zhou, D.; Gao, J. Metagenomic analysis exploring microbial assemblages and functional genes potentially involved in di (2-ethylhexyl) phthalate degradation in soil. Sci. Total Environ. 2020, 715, 137037. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.; Lindeque, P.; Fileman, E.; Halsband, C.; Goodhead, R.; Moger, J.; Galloway, T.S. Microplastic ingestion by zooplankton. Environ. Sci. Technol. 2013, 47, 6646–6655. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.L.; Stanton, T.; Law, A. Plastic habitats: Algal biofilms on photic and aphotic plastics. J. Hazard. Mater. Lett. 2021, 2, 100038. [Google Scholar] [CrossRef]
- Canniff, P.M.; Hoang, T.C. Microplastic ingestion by Daphnia magna and its enhancement on algal growth. Sci. Total Environ. 2018, 633, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Besseling, E.; Wegner, A.; Foekema, E.M.; van den Heuvel-Greve, M.J.; Koelmans, A.A. Effects of microplastic on fitness and PCB bioaccumulation by the lugworm Arenicola marina (L.). Environ. Sci. Technol. 2013, 47, 593–600. [Google Scholar] [CrossRef]
- Kokkuar, N.; Li, L.; Srisapoome, P.; Dong, S.; Tian, X. Application of biodegradable polymers as carbon sources in ex situ biofloc systems: Water quality and shift of microbial community. Aquacult. Res. 2021, 52, 3570–3579. [Google Scholar] [CrossRef]
- Ling, J.; Zhou, W.; Yang, Q.; Yin, J.; Zhang, J.; Peng, Q.; Huang, X.; Zhang, Y.; Dong, J. Spatial and species variations of bacterial community structure and putative function in seagrass rhizosphere sediment. Life 2021, 11, 852. [Google Scholar] [CrossRef]
- Ugwu, J.A.; Wenzi, R.; Asiegbu, F.O. Monocot diet sources drive diversity of gut bacterial communities in Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae. J. Appl. Entomol. 2022, 146, 942–956. [Google Scholar] [CrossRef]
- Tang, C.; Yi, Y.; Yang, Z.; Zhou, Y.; Zerizghi, T.; Wang, X.; Cui, X.; Duan, P. Planktonic indicators of trophic states for a shallow lake (Baiyangdian Lake, China). Limnologica 2019, 78, 125712. [Google Scholar] [CrossRef]
- Lv, P.; Luo, J.; Zhuang, X.; Zhang, D.; Huang, Z.; Bai, Z. Diversity of culturable aerobic denitrifying bacteria in the sediment, water and biofilms in Liangshui River of Beijing, China. Sci. Rep. 2017, 7, 10032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, P.; Yang, D.; Shan, L.; Wang, D.; Li, Q.; Gorski, L.; Lee, B.G.; Quiñones, B.; Cooley, M.B. Concurrent detection of human norovirus and bacterial pathogens in water samples from an agricultural region in central California coast. Front. Microbiol. 2017, 8, 1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, C.; Reckhow, K.H.; Lukasik, J.; Harwood, V.J. Assessment of sources of human pathogens and fecal contamination in a Florida freshwater lake. Water Res. 2012, 46, 5799–5812. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.E. Viral pathogens in water: Occurrence, public health impact, and available control strategies. Curr. Opin. Virol. 2014, 4, 50–57. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Estimators | AB (Mean ± SD) | NB (Mean ± SD) | p Value |
|---|---|---|---|
| Shannon | 1.86 ± 0.83 | 4.27 ± 0.41 | <0.001 |
| Chao | 237.26 ± 114.92 | 769.47 ± 188.45 | <0.001 |
| Coverage | 0.99 ± 0.01 | 0.97 ± 0.01 | <0.001 |
| Pd | 26.48 ± 9.02 | 57.69 ± 11.80 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Feng, S.; Gao, F.; Wen, H.; Zhu, L.; Li, M.; Xi, Y.; Xiang, X. The Relationship between Brachionus calyciflorus-Associated Bacterial and Bacterioplankton Communities in a Subtropical Freshwater Lake. Animals 2022, 12, 3201. https://doi.org/10.3390/ani12223201
Zhang Y, Feng S, Gao F, Wen H, Zhu L, Li M, Xi Y, Xiang X. The Relationship between Brachionus calyciflorus-Associated Bacterial and Bacterioplankton Communities in a Subtropical Freshwater Lake. Animals. 2022; 12(22):3201. https://doi.org/10.3390/ani12223201
Chicago/Turabian StyleZhang, Yongzhi, Sen Feng, Fan Gao, Hao Wen, Lingyun Zhu, Meng Li, Yilong Xi, and Xianling Xiang. 2022. "The Relationship between Brachionus calyciflorus-Associated Bacterial and Bacterioplankton Communities in a Subtropical Freshwater Lake" Animals 12, no. 22: 3201. https://doi.org/10.3390/ani12223201