
Rumen Microbial Predictors for Short-Chain Fatty Acid Levels and the Grass-Fed Regimen in Angus Cattle
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. Measurements of Short-Chain Fatty Acids and Lactate
2.3. DNA Extraction and 16S rRNA Gene Sequencing
2.4. Bioinformatics and Statistical Analyses
3. Results
3.1. The Differences in Rumen Microbial Composition between Grass-Fed and Grain-Fed Cattle
3.2. Microbial Taxa Significantly Correlated with Rumen Short-Chain Fatty Acid Contents
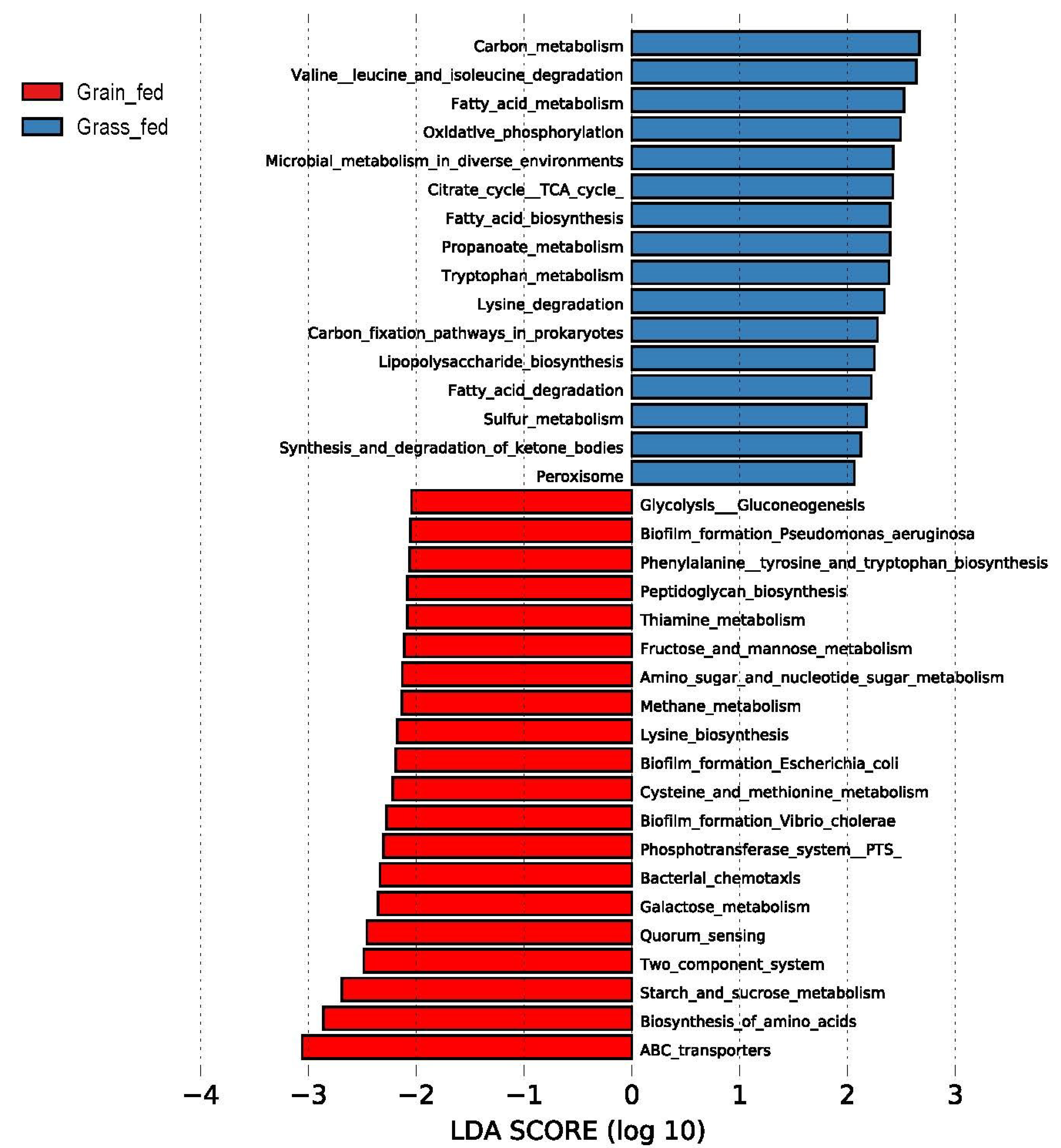
3.3. Differentially Abundant Taxa and Predicted Function Categories between the Two Feeding Regimens
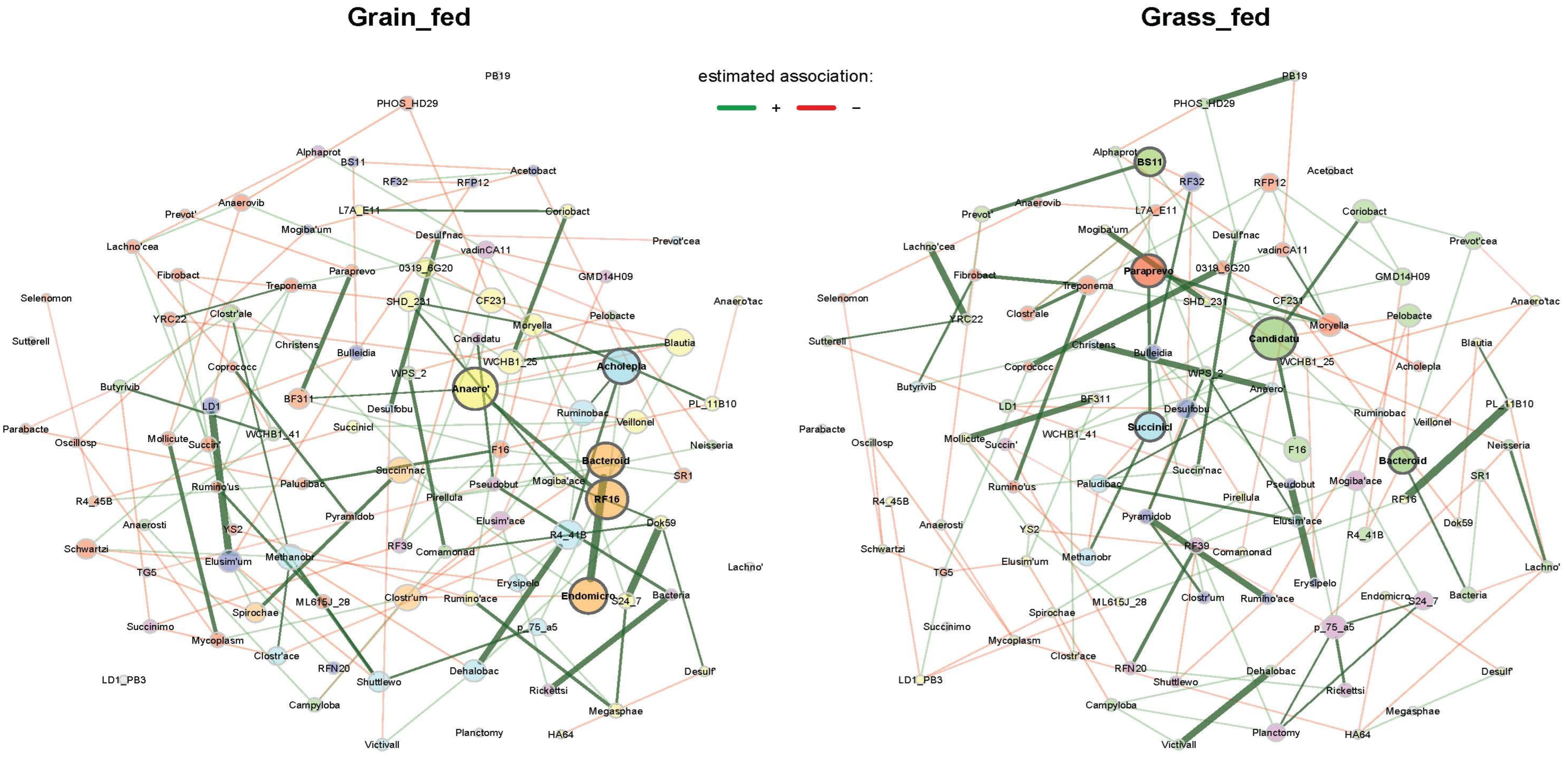
3.4. Rumen Microbial Interaction Networks
3.5. Identification of Microbial Predictors or Balances Using the Selbal Algorithm
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackie, R.I. Mutualistic fermentative digestion in the gastrointestinal tract: Diversity and evolution. Integr. Comp. Biol. 2002, 42, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, P.J. Nutritional Ecology of the Ruminant, 2nd ed.; Comstock Pub.: Ithaca, NY, USA, 1994. [Google Scholar]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannink, A.; Kogut, J.; Dijkstra, J.; France, J.; Kebreab, E.; Van Vuuren, A.M.; Tamminga, S. Estimation of the stoichiometry of volatile fatty acid production in the rumen of lactating cows. J. Theor. Biol. 2006, 238, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Moran, J. Tropical Dairy Farming: Feeding Management for Small Holder Dairy Farmers in the Humid Tropics; Land Links: Collingwood, VIC, Australia, 2005. [Google Scholar]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin, V.; Ransom, L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, T.G.; Titgemeyer, E.C. Ruminal acidosis in beef cattle: The current microbiological and nutritional outlook. J. Dairy Sci. 2007, 90 (Suppl. S1), E17–E38. [Google Scholar] [CrossRef] [Green Version]
- Daley, C.A.; Abbott, A.; Doyle, P.S.; Nader, G.A.; Larson, S. A review of fatty acid profiles and antioxidant content in grass-fed and grain-fed beef. Nutr. J. 2010, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, J.A.; Bai, Y.; He, Y.; Li, Y.; Cai, W.; Bickhart, D.M.; Liu, G.; Barao, S.M.; Sonstegard, T.; Song, J. Growth curve, blood parameters and carcass traits of grass-fed Angus steers. Animal 2021, 15, 100381. [Google Scholar] [CrossRef]
- Liu, J.; Liu, F.; Cai, W.; Jia, C.; Bai, Y.; He, Y.; Zhu, W.; Li, R.W.; Song, J. Diet-induced changes in bacterial communities in the jejunum and their associations with bile acids in Angus beef cattle. Anim. Microbiome 2020, 2, 1–14. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Peschel, S.; Müller, C.L.; von Mutius, E.; Boulesteix, A.-L.; Depner, M. NetCoMi: Network construction and comparison for microbiome data in R. Brief. Bioinform. 2021, 22, bbaa290. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Gaynanova, I.; Muller, C.L. Microbial Networks in SPRING—Semi-parametric Rank-Based Correlation and Partial Correlation Estimation for Quantitative Microbiome Data. Front. Genet. 2019, 10, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Przemieniecki, S.W.; Kosewska, A.; Ciesielski, S.; Kosewska, O. Changes in the gut microbiome and enzymatic profile of Tenebrio molitor larvae biodegrading cellulose, polyethylene and polystyrene waste. Environ. Pollut. 2020, 256, 113265. [Google Scholar] [CrossRef]
- Solden, L.M.; Hoyt, D.W.; Collins, W.B.; Plank, J.E.; Daly, R.A.; Hildebrand, E.; Beavers, T.J.; Wolfe, R.; Nicora, C.D.; Purvine, S.O.; et al. New roles in hemicellulosic sugar fermentation for the uncultivated Bacteroidetes family BS11. ISME J. 2017, 11, 691–703. [Google Scholar] [CrossRef]
- Downes, J.; Vartoukian, S.R.; Dewhirst, F.E.; Izard, J.; Chen, T.; Yu, W.H.; Sutcliffe, I.C.; Wade, W.G. Pyramidobacter piscolens gen. nov., sp. nov., a member of the phylum ‘Synergistetes’ isolated from the human oral cavity. Int. J. Syst. Evol. Microbiol. 2009, 59, 972–980. [Google Scholar] [CrossRef]
- Hayek, M.N.; Garrett, R.D. Nationwide shift to grass-fed beef requires larger cattle population. Environ. Res. Lett. 2018, 13, 084005. [Google Scholar] [CrossRef]
- Oliveira, P.P.A.; Berndt, A.; Pedroso, A.F.; Alves, T.C.; Pezzopane, J.R.M.; Sakamoto, L.S.; Henrique, F.L.; Rodrigues, P.H.M. Greenhouse gas balance and carbon footprint of pasture-based beef cattle production systems in the tropical region (Atlantic Forest biome). Animal 2020, 14, s427–s437. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.G.N.; Bressani, F.A.; Cuadrat, R.R.C.; Cardoso, T.F.; Malheiros, J.M.; de Oliveira, P.S.N.; Petrini, J.; Mourao, G.B.; Coutinho, L.L.; Reecy, J.M.; et al. Stool and Ruminal Microbiome Components Associated With Methane Emission and Feed Efficiency in Nelore Beef Cattle. Front. Genet. 2022, 13, 812828. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Misselbrook, T.H.; Vega, A.; Gonzalez-Quintero, R.; Chavarro-Lobo, J.A.; Mazzetto, A.M.; Chadwick, D.R. Measured ammonia emissions from tropical and subtropical pastures: A comparison with 2006 IPCC, 2019 Refinement to the 2006 IPCC, and EMEP/EEA (European Monitoring and Evaluation Programme and European Environmental Agency) inventory estimates. J. Dairy Sci. 2020, 103, 6706–6715. [Google Scholar] [CrossRef] [PubMed]
- AlZahal, O.; Li, F.; Guan, L.L.; Walker, N.D.; McBride, B.W. Factors influencing ruminal bacterial community diversity and composition and microbial fibrolytic enzyme abundance in lactating dairy cows with a focus on the role of active dry yeast. J. Dairy Sci. 2017, 100, 4377–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siciliano-Jones, J.; Murphy, M.R. Production of volatile fatty acids in the rumen and cecum-colon of steers as affected by forage:concentrate and forage physical form. J. Dairy Sci. 1989, 72, 485–492. [Google Scholar] [CrossRef]
- Pichler, M.J.; Yamada, C.; Shuoker, B.; Alvarez-Silva, C.; Gotoh, A.; Leth, M.L.; Schoof, E.; Katoh, T.; Sakanaka, M.; Katayama, T.; et al. Butyrate producing colonic Clostridiales metabolise human milk oligosaccharides and cross feed on mucin via conserved pathways. Nat. Commun. 2020, 11, 3285. [Google Scholar] [CrossRef]
- Ferrario, C.; Taverniti, V.; Milani, C.; Fiore, W.; Laureati, M.; De Noni, I.; Stuknyte, M.; Chouaia, B.; Riso, P.; Guglielmetti, S. Modulation of fecal Clostridiales bacteria and butyrate by probiotic intervention with Lactobacillus paracasei DG varies among healthy adults. J. Nutr. 2014, 144, 1787–1796. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shen, Y.; Wang, C.; Ding, L.; Zhao, F.; Wang, M.; Fu, J.; Wang, H. Megasphaera elsdenii Lactate Degradation Pattern Shifts in Rumen Acidosis Models. Front. Microbiol. 2019, 10, 162. [Google Scholar] [CrossRef] [Green Version]
- Dedysh, S.N.; Kulichevskaya, I.S.; Beletsky, A.V.; Ivanova, A.A.; Rijpstra, W.I.C.; Damsté, J.S.S.; Mardanov, A.V.; Ravin, N.V. Lacipirellula parvula gen. nov., sp. nov., representing a lineage of planctomycetes widespread in low-oxygen habitats, description of the family Lacipirellulaceae fam. nov. and proposal of the orders Pirellulales ord. nov., Gemmatales ord. nov. and Isosphaerales ord. nov. Syst. Appl. Microbiol. 2020, 43, 126050. [Google Scholar] [CrossRef]
- Goad, D.W.; Goad, C.L.; Nagaraja, T.G. Ruminal microbial and fermentative changes associated with experimentally induced subacute acidosis in steers. J. Anim. Sci. 1998, 76, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Pope, P.B.; Smith, W.; Denman, S.E.; Tringe, S.G.; Barry, K.; Hugenholtz, P.; McSweeney, C.S.; McHardy, A.C.; Morrison, M. Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar wallabies. Science 2011, 333, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Indugu, N.; Vecchiarelli, B.; Baker, L.D.; Ferguson, J.D.; Vanamala, J.K.P.; Pitta, D.W. Comparison of rumen bacterial communities in dairy herds of different production. BMC Microbiol. 2017, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 2013, 79, 3744–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Section, F.B.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [Green Version]
- Romero-Perez, G.A.; Ominski, K.H.; McAllister, T.A.; Krause, D.O. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl. Environ. Microbiol. 2011, 77, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.Y.; Ye, H.M.; Liu, J.H.; Mao, S.Y. High-grain diets altered rumen fermentation and epithelial bacterial community and resulted in rumen epithelial injuries of goats. Appl. Microbiol. Biot. 2017, 101, 6981–6992. [Google Scholar] [CrossRef]
- Gozho, G.N.; Krause, D.O.; Plaizier, J.C. Rumen lipopolysaccharide and inflammation during grain adaptation and subacute ruminal acidosis in steers. J. Dairy Sci. 2006, 89, 4404–4413. [Google Scholar] [CrossRef] [Green Version]
- Urrutia, N.L.; Harvatine, K.J. Acetate Dose-Dependently Stimulates Milk Fat Synthesis in Lactating Dairy Cows. J. Nutr. 2017, 147, 763–769. [Google Scholar] [CrossRef] [Green Version]
- Yost, W.M.; Young, J.W.; Schmidt, S.P.; McGilliard, A.D. Gluconeogenesis in ruminants: Propionic acid production from a high-grain diet fed to cattle. J. Nutr. 1977, 107, 2036–2043. [Google Scholar] [CrossRef]
- Russell, J.B. The importance of pH in the regulation of ruminal acetate to propionate ratio and methane production in vitro. J. Dairy Sci. 1998, 81, 3222–3230. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VFA | Grain-Fed | Grass-Fed | p Value |
|---|---|---|---|
| Acetate | 3990.125 ± 420.756 | 3197.556 ± 883.497 | 0.0338 * |
| Propionate | 1515.625 ± 214.071 | 834.889 ± 296.516 | <0.0001 *** |
| Butyrate | 773.125 ± 90.044 | 539.444 ± 200.4 | 0.0088 ** |
| Isobutyrate | 119.5 ± 10.61 | 69.111 ± 8.638 | <0.0001 *** |
| Lactic acid | 8.286 ± 6.264 | 2.667 ± 3.162 | 0.0605 . |
| Grain-Fed | Grass-Fed | Abs.Diff. | p Value | |
|---|---|---|---|---|
| Global network measures: | ||||
| Average path length | 2.761 | 2.863 | 0.102 | 0.527473 |
| Clustering coefficient | 0.052 | 0.12 | 0.067 | 0.074925 . |
| Modularity | 0.494 | 0.586 | 0.092 | 0.002997 ** |
| Vertex connectivity | 1 | 1 | 0 | 1 |
| Edge connectivity | 1 | 1 | 0 | 1 |
| Edge density | 0.038 | 0.037 | 0.001 | 0.818182 |
| Degree (normalized): | ||||
| Lachnospira | 0 | 0.061 | 0.061 | 0.620445 |
| Candidatus Amoebophilus | 0.03 | 0.081 | 0.051 | 0.620445 |
| Unclassified_LD1-PB3 | 0 | 0.051 | 0.051 | 0.620445 |
| Unclassified_Planctomycetes | 0 | 0.04 | 0.04 | 0.620445 |
| Acetobacter | 0.04 | 0 | 0.04 | 0.802088 |
| Desulfobulbus | 0.03 | 0.071 | 0.04 | 0.620445 |
| Unclassified_Lachnospiraceae | 0.051 | 0.01 | 0.04 | 0.620445 |
| Anaeroplasma | 0.071 | 0.03 | 0.04 | 0.620445 |
| Clostridium | 0.061 | 0.02 | 0.04 | 0.620445 |
| SHD-231 | 0.051 | 0.01 | 0.04 | 0.620445 |
| Eigenvector centrality (normalized): | ||||
| Anaeroplasma | 1 | 0.09 | 0.91 | 0.097113 . |
| Unclassified_RF16 | 0.949 | 0.058 | 0.891 | 0.218505 |
| Candidatus Amoebophilus | 0.166 | 1 | 0.834 | 0.58268 |
| Unclassified_Acholeplasmatales | 0.928 | 0.098 | 0.831 | 0.097113 . |
| Unclassified_Endomicrobia | 0.72 | 0.017 | 0.703 | 0.097113 . |
| Unclassified_RFP12 | 0.132 | 0.701 | 0.569 | 0.776907 |
| Clostridium | 0.575 | 0.056 | 0.519 | 0.49944 |
| Blautia | 0.515 | 0.026 | 0.489 | 0.49944 |
| Treponema | 0.171 | 0.659 | 0.488 | 0.794249 |
| Unclassified_Veillonellaceae | 0.501 | 0.014 | 0.488 | 0.49944 |
| j | p (J ≤ j) | p (J ≥ j) | |
|---|---|---|---|
| degree | 0.088 | 0.000934 *** | 0.999837 |
| betweenness centr. | 0.19 | 0.031715 * | 0.986817 |
| closeness centr. | 0.19 | 0.031715 * | 0.986817 |
| eigenvec. centr. | 0.163 | 0.010359 * | 0.996387 |
| hub taxa | 0.111 | 0.143068 | 0.973988 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Bai, Y.; Liu, F.; Kohn, R.A.; Tadesse, D.A.; Sarria, S.; Li, R.W.; Song, J. Rumen Microbial Predictors for Short-Chain Fatty Acid Levels and the Grass-Fed Regimen in Angus Cattle. Animals 2022, 12, 2995. https://doi.org/10.3390/ani12212995
Liu J, Bai Y, Liu F, Kohn RA, Tadesse DA, Sarria S, Li RW, Song J. Rumen Microbial Predictors for Short-Chain Fatty Acid Levels and the Grass-Fed Regimen in Angus Cattle. Animals. 2022; 12(21):2995. https://doi.org/10.3390/ani12212995
Chicago/Turabian StyleLiu, Jianan, Ying Bai, Fang Liu, Richard A. Kohn, Daniel A. Tadesse, Saul Sarria, Robert W. Li, and Jiuzhou Song. 2022. "Rumen Microbial Predictors for Short-Chain Fatty Acid Levels and the Grass-Fed Regimen in Angus Cattle" Animals 12, no. 21: 2995. https://doi.org/10.3390/ani12212995