

Complete Mitogenomes of Polypedates Tree Frogs Unveil Gene Rearrangement and Concerted Evolution within Rhacophoridae
 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. WGS Library Preparation
2.3. Genome Assembly and LA-PCR Amplification
2.4. Mitogenome Annotation
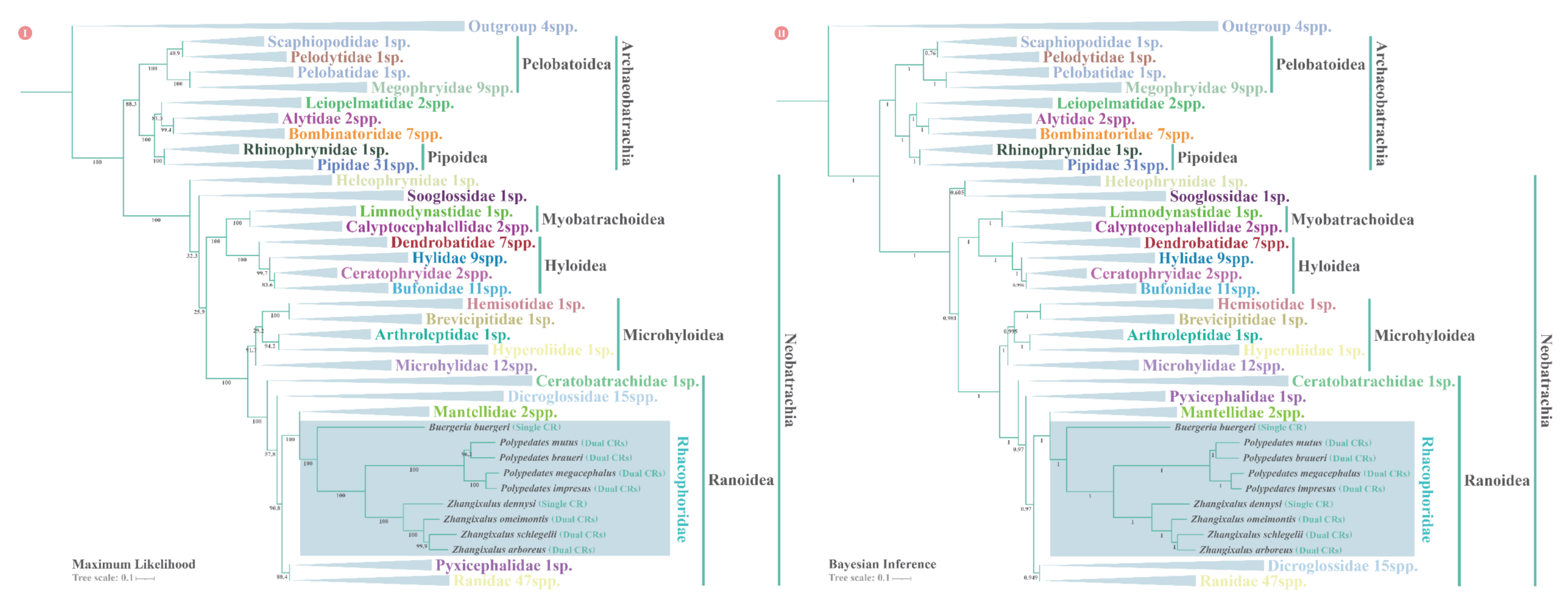
2.5. Phylogenetic Analyses
3. Results
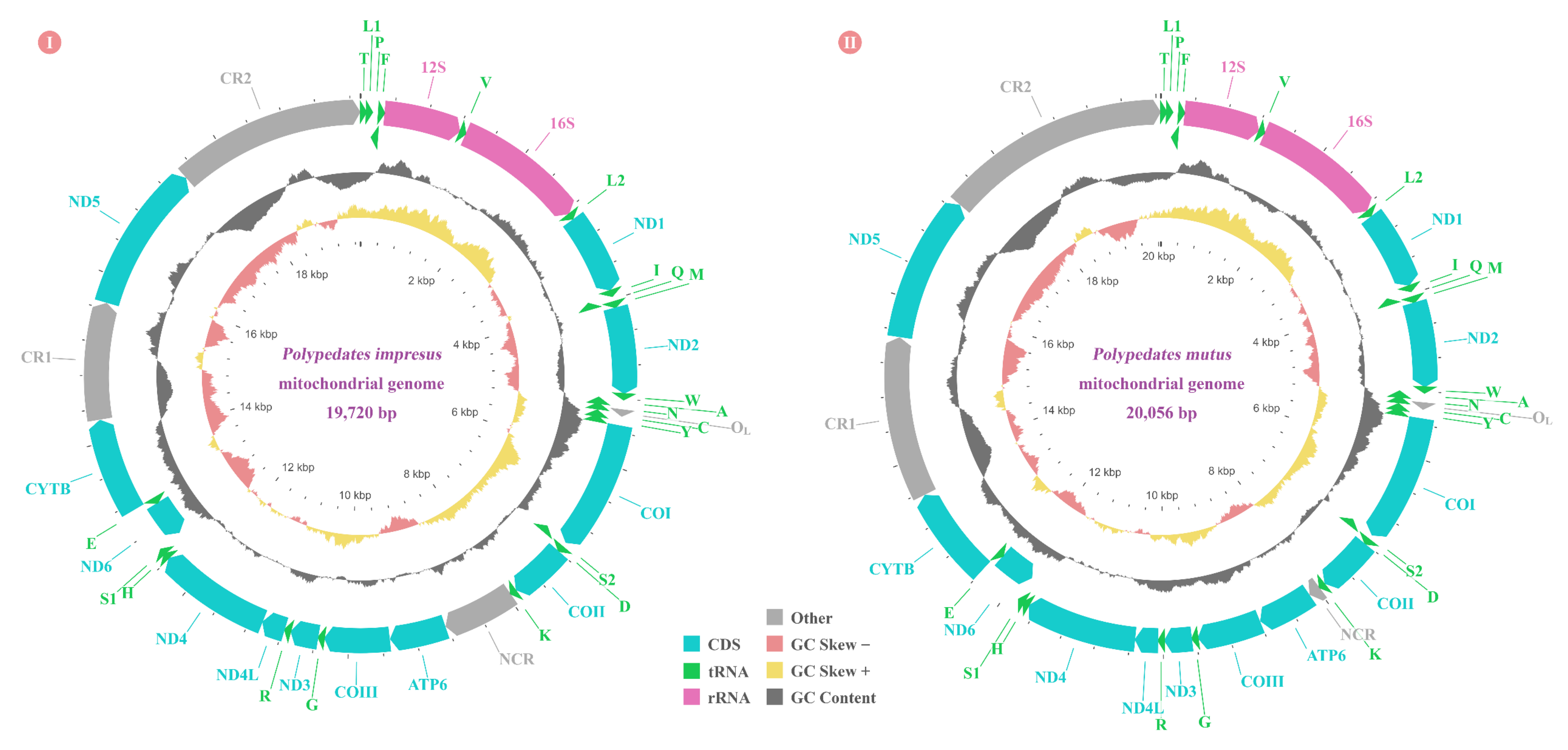
3.1. Mitogenome Organization and Nucleotide Composition
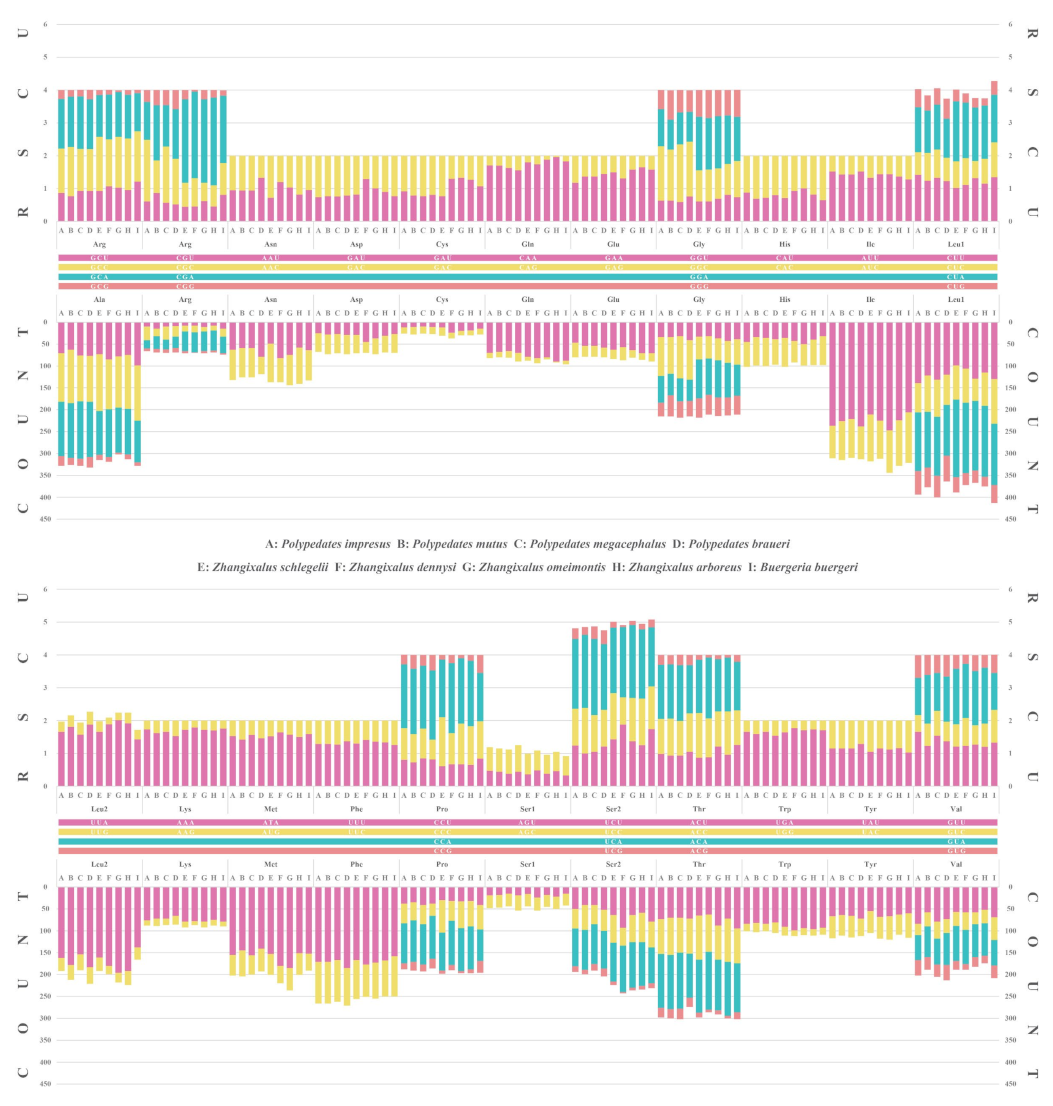
3.2. Protein-Coding Genes and Codon Usage
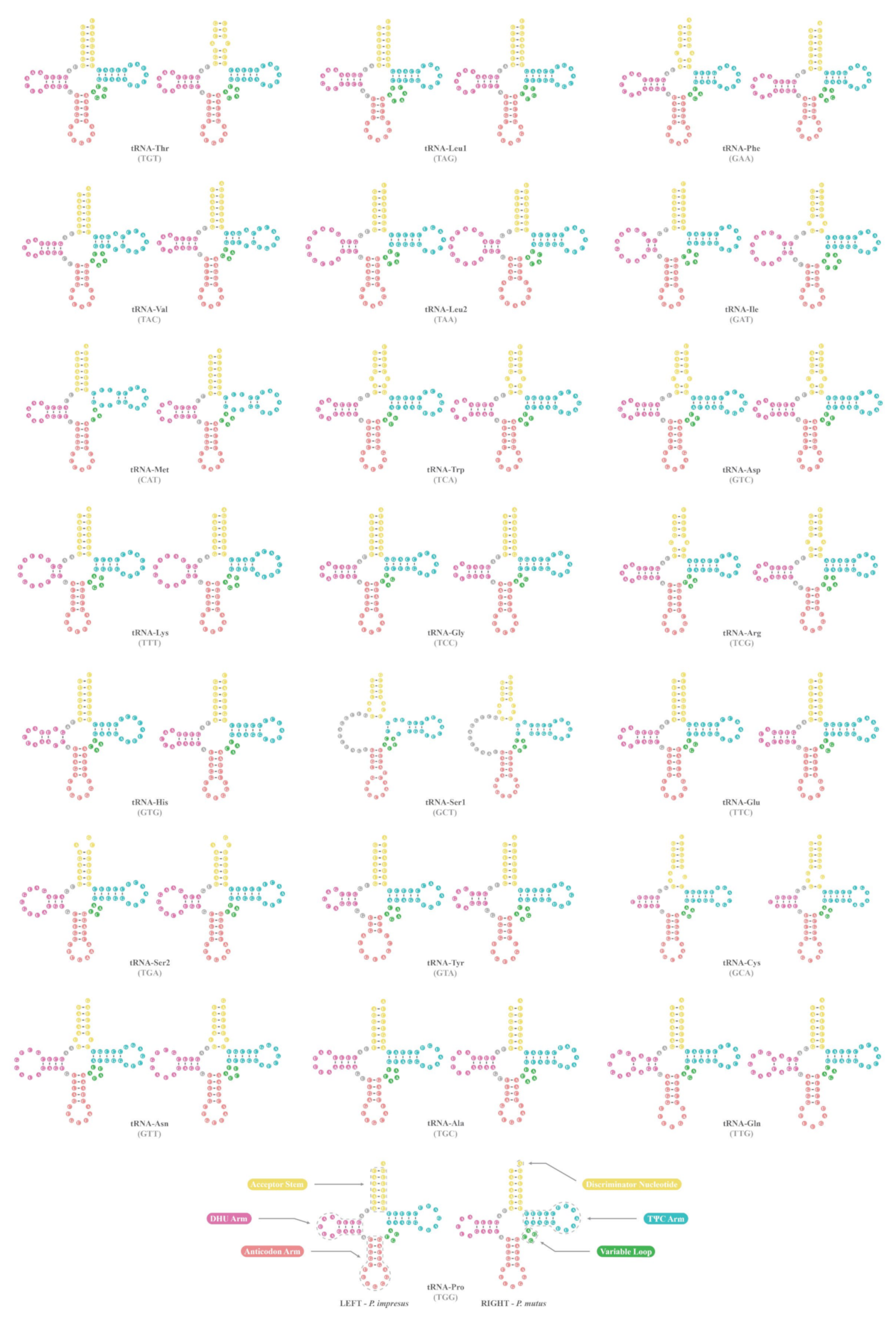
3.3. Transfer and Ribosomal RNA Genes
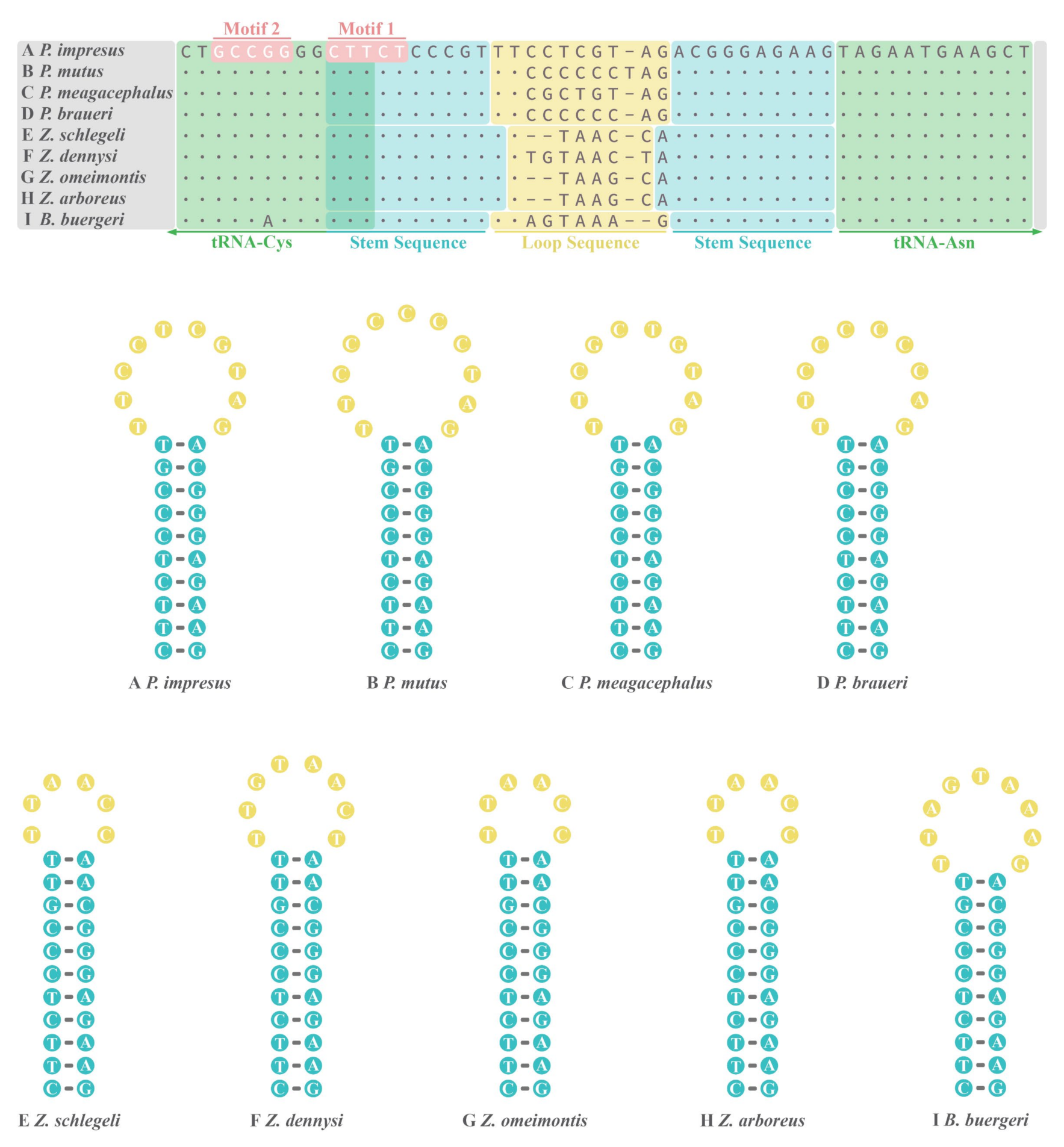
3.4. OL of Nine Rhacophorids
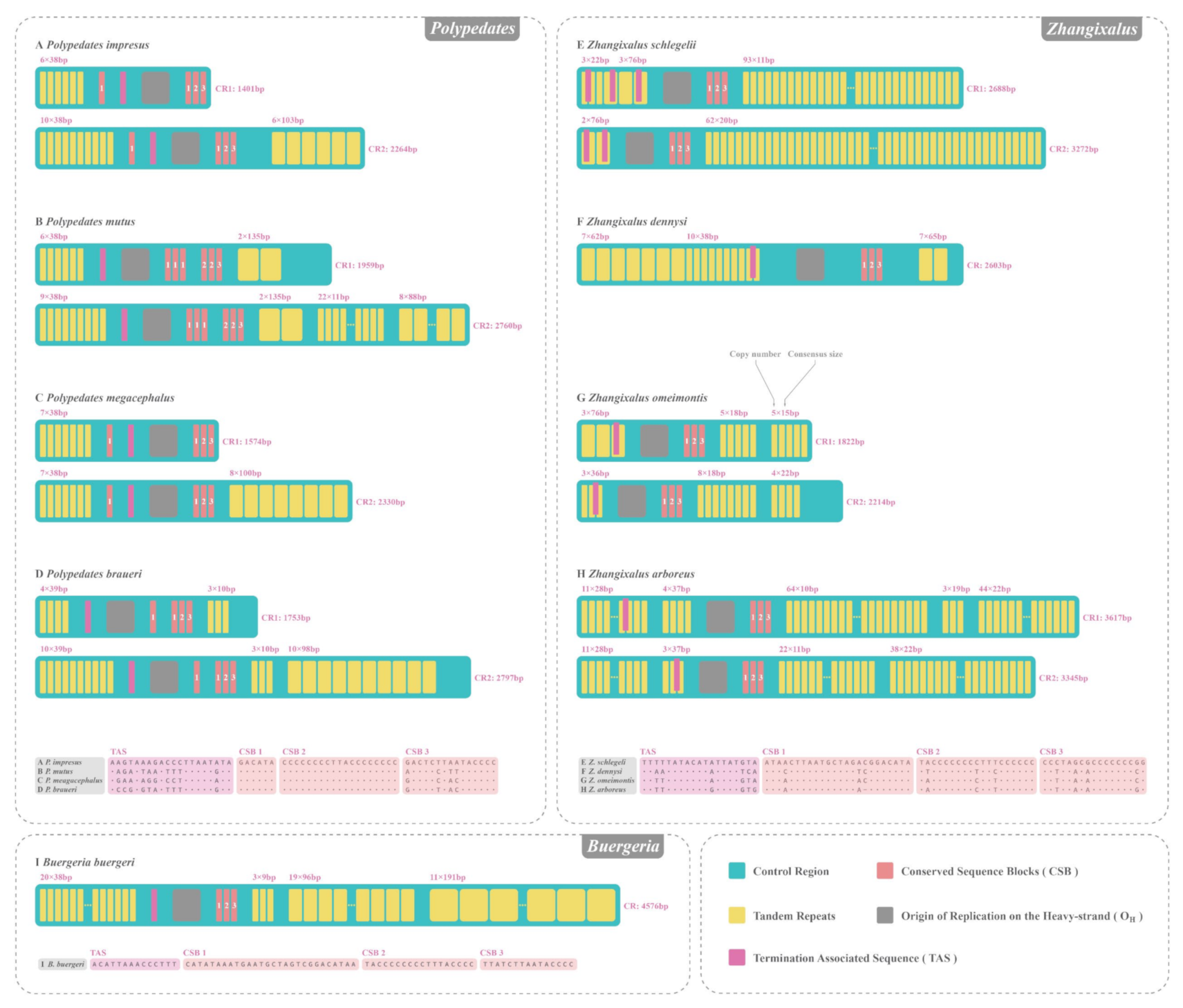
3.5. CR of Nine Rhacophorids
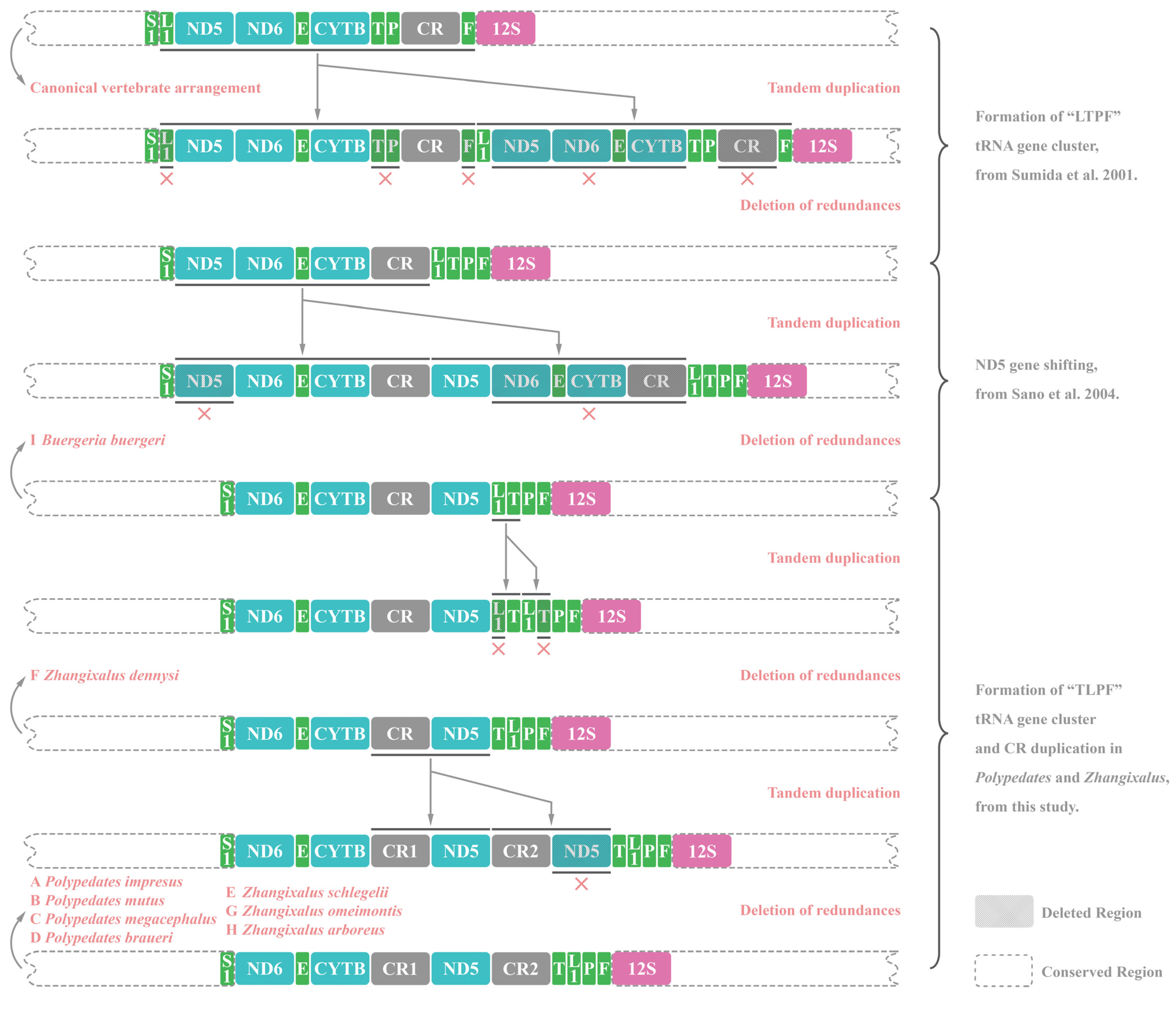
3.6. Gene Rearrangements of Nine Rhacophorids
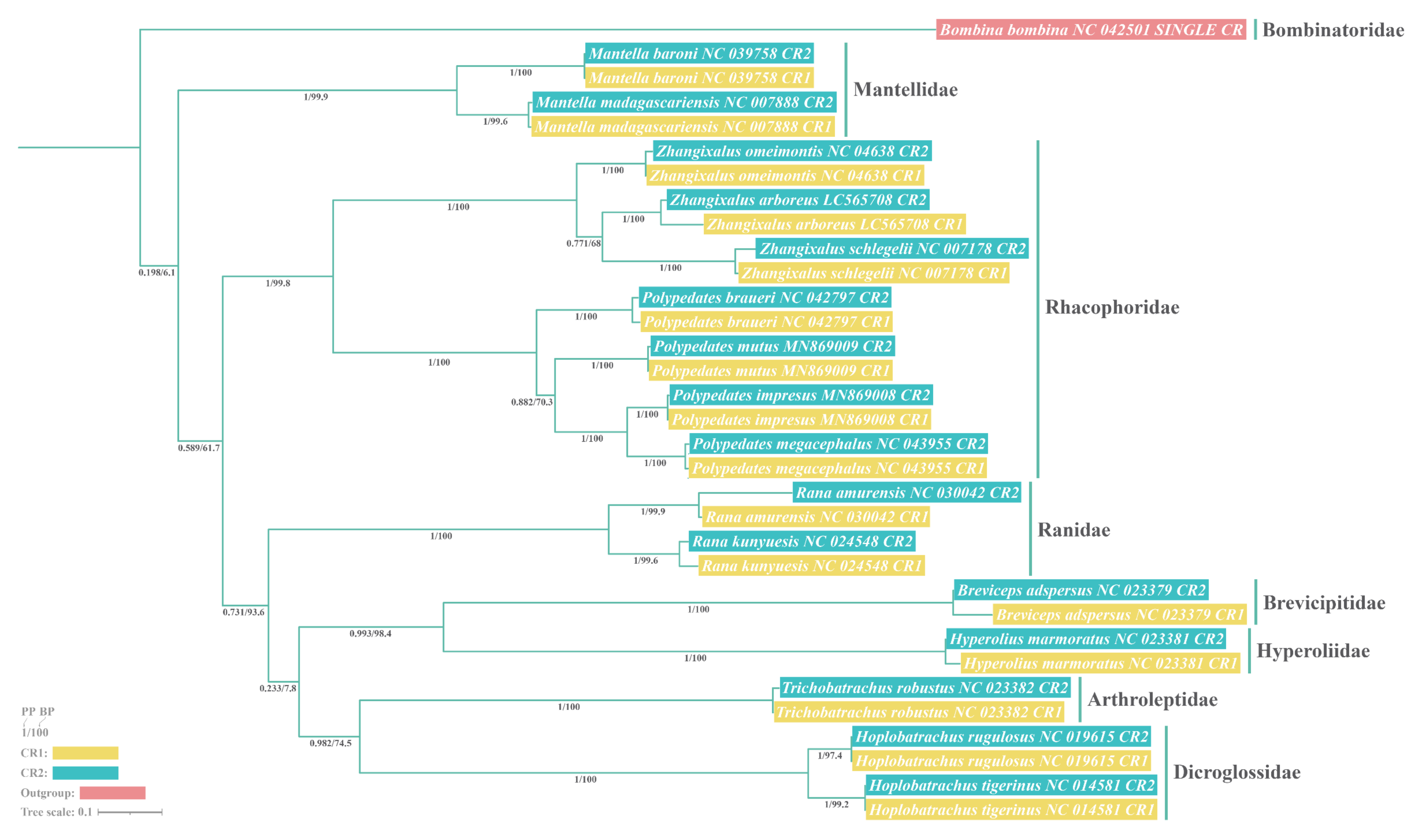
3.7. Phylogenetic Relationships
4. Discussion
4.1. Mitogenome Sructural Analyses of Nine Rhacophorids
4.2. CR Sructural Analyses of Nine Rhacophorids
4.3. Concerted Evolution of the CRs in Nine Rhacophorids
4.4. Causes and Advantages of Dual CRs
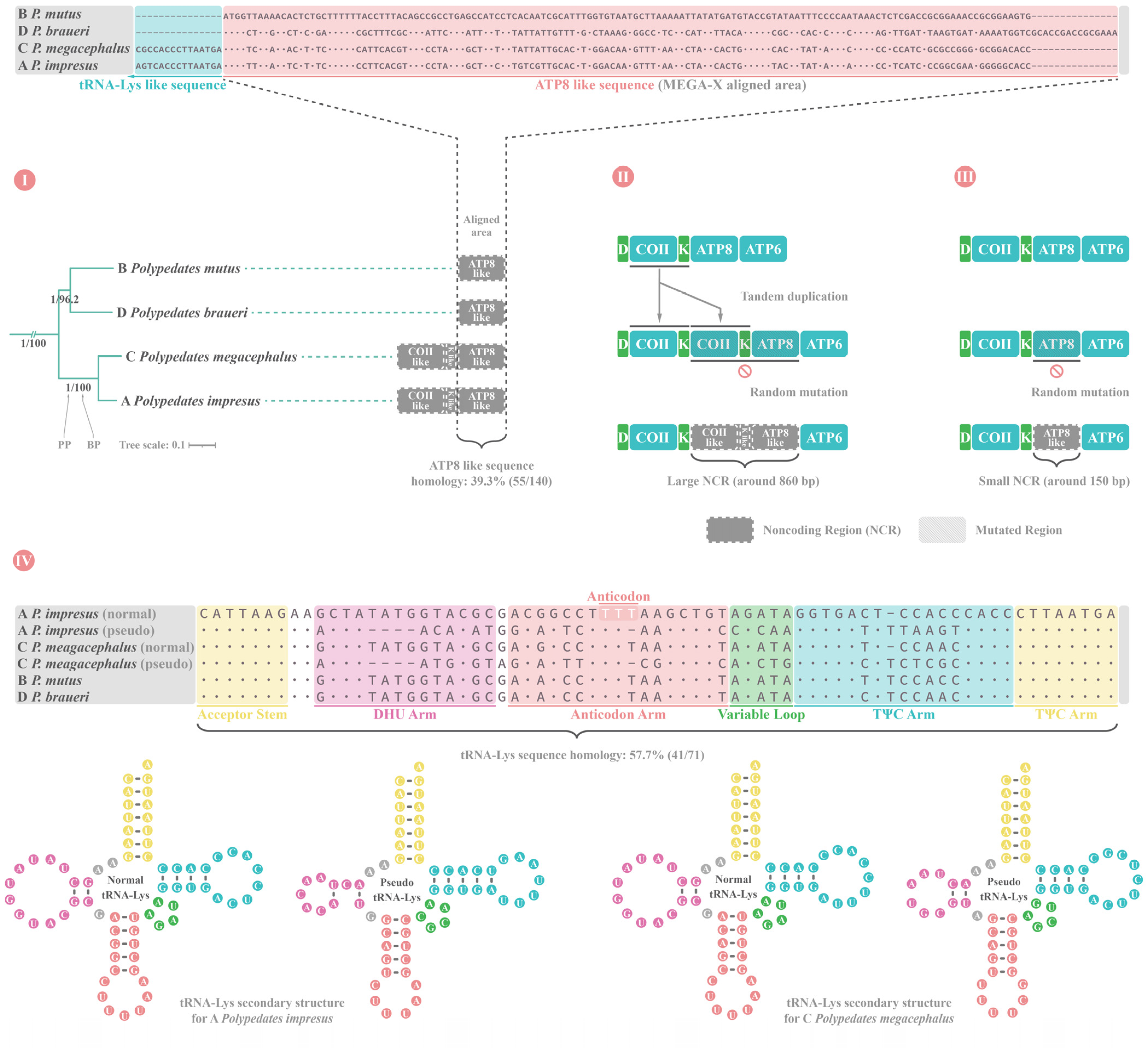
4.5. Parallel Evolution of ATP8 Gene LoF in Four Polypedates and CR Duplication in Nine Rhacophorids
4.6. Phylogenetic Analyses and Its Insight into Gene Rearrangements of Nine Rhacophorids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Hjort, K.; Goldberg, A.V.; Tsaousis, A.D.; Hirt, R.P.; Embley, T.M. Diversity and reductive evolution of mitochondria among microbial eukaryotes. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Vanhove, M.P.; Briscoe, A.G.; Jorissen, M.W.; Littlewood, D.T.J.; Huyse, T. The first next-generation sequencing approach to the mitochondrial phylogeny of African monogenean parasites (Platyhelminthes: Gyrodactylidae and Dactylogyridae). BMC Genom. 2018, 19, 520. [Google Scholar] [CrossRef]
- Ramos, B.; González-Acuña, D.; Loyola, D.E.; Johnson, W.E.; Parker, P.G.; Massaro, M.; Dantas, G.P.; Miranda, M.D.; Vianna, J.A. Landscape genomics: Natural selection drives the evolution of mitogenome in penguins. BMC Genom. 2018, 19, 53. [Google Scholar] [CrossRef]
- Sharma, A.; Siva, C.; Ali, S.; Sahoo, P.K.; Nath, R.; Laskar, M.; Sarma, D. The complete mitochondrial genome of the medicinal fish, Cyprinion semiplotum: Insight into its structural features and phylogenetic implications. Int. J. Biol. Macromol. 2020, 164, 939–948. [Google Scholar] [CrossRef]
- Jiang, L.; Zhao, L.; Cheng, D.; Zhu, L.; Zhang, M.; Ruan, Q.; Chen, W. The complete mitochondrial genome sequence of the Sichuan Digging Frog, Kaloula rugifera (Anura: Microhylidae) and its phylogenetic implications. Gene 2017, 626, 367–375. [Google Scholar] [CrossRef]
- American Museum of Natural History. Amphibian Species of the World: An Online Reference, Version 6.1. Available online: https://amphibiansoftheworld.amnh.org/index.php (accessed on 28 October 2021).
- Chen, J.-M.; Prendini, E.; Wu, Y.-H.; Zhang, B.-L.; Suwannapoom, C.; Chen, H.-M.; Jin, J.-Q.; Lemmon, E.M.; Lemmon, A.R.; Stuart, B.L. An integrative phylogenomic approach illuminates the evolutionary history of Old World tree frogs (Anura: Rhacophoridae). Mol. Phylogenet. Evol. 2020, 145, 106724. [Google Scholar] [CrossRef]
- Meegaskumbura, M.; Senevirathne, G.; Biju, S.; Garg, S.; Meegaskumbura, S.; Pethiyagoda, R.; Hanken, J.; Schneider, C.J. Patterns of reproductive-mode evolution in Old World tree frogs (Anura, Rhacophoridae). Zool. Scr. 2015, 44, 509–522. [Google Scholar] [CrossRef]
- Feng, Y.-J.; Blackburn, D.C.; Liang, D.; Hillis, D.M.; Wake, D.B.; Cannatella, D.C.; Zhang, P. Phylogenomics reveals rapid, simultaneous diversification of three major clades of Gondwanan frogs at the Cretaceous–Paleogene boundary. Proc. Natl. Acad. Sci. USA 2017, 114, E5864–E5870. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.M.; Linkem, C.W.; Siler, C.D.; Sukumaran, J.; Esselstyn, J.A.; Diesmos, A.C.; Iskandar, D.T.; Bickford, D.; Evans, B.J.; McGuire, J.A. Phylogeography and historical demography of Polypedates leucomystax in the islands of Indonesia and the Philippines: Evidence for recent human-mediated range expansion? Mol. Phylogenet. Evol. 2010, 57, 598–619. [Google Scholar] [CrossRef]
- Kuraishi, N.; Matsui, M.; Hamidy, A.; Belabut, D.M.; Ahmad, N.; Panha, S.; Sudin, A.; Yong, H.S.; Jiang, J.P.; Ota, H. Phylogenetic and taxonomic relationships of the Polypedates leucomystax complex (Amphibia). Zool. Scr. 2013, 42, 54–70. [Google Scholar] [CrossRef]
- Pan, S.; Dang, N.; Wang, J.; Zheng, Y.; Rao, D.; Li, J. Molecular phylogeny supports the validity of Polypedates impresus Yang 2008. Asian Herpetol. Res. 2013, 4, 124–133. [Google Scholar]
- Amphibia China; Kunming Institute of Zoology (CAS). The Database of Chinese Amphibians. Available online: http://www.amphibiachina.org/ (accessed on 28 October 2021).
- Alam, M.S.; Kurabayashi, A.; Hayashi, Y.; Sano, N.; Khan, M.M.R.; Fujii, T.; Sumida, M. Complete mitochondrial genomes and novel gene rearrangements in two dicroglossid frogs, Hoplobatrachus tigerinus and Euphlyctis hexadactylus, from Bangladesh. Genes Genet. Syst. 2010, 85, 219–232. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, J.; Zheng, R.; Shao, C. The complete mitochondrial genome of Hoplobatrachus rugulosus (Anura: Dicroglossidae). Mitochondrial DNA 2012, 23, 336–337. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Usuki, C.; Mikami, N.; Fujii, T.; Yonekawa, H.; Sumida, M.; Hasegawa, M. Complete nucleotide sequence of the mitochondrial genome of a Malagasy poison frog Mantella madagascariensis: Evolutionary implications on mitochondrial genomes of higher anuran groups. Mol. Phylogenet. Evol. 2006, 39, 223–236. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef]
- Sano, N.; Kurabayashi, A.; Fujii, T.; Yonekawa, H.; Sumida, M. Complete nucleotide sequence of the mitochondrial genome of Schlegel’s tree frog Rhacophorus schlegelii (family Rhacophoridae): Duplicated control regions and gene rearrangements. Genes Genet. Syst. 2005, 80, 213–224. [Google Scholar] [CrossRef]
- Huang, A.; Li, H.; Luo, H.; Ni, Q.; Yao, Y.; Xu, H.; Zeng, B.; Li, Y.; Wei, Z.; Zhang, M. The complete mitochondrial genome of the tree frog, Polypedates braueri (Anura, Rhacophoridae). Mitochondrial DNA Part B 2019, 4, 1739–1740. [Google Scholar] [CrossRef]
- Huang, A.; Liu, S.; Li, H.; Luo, H.; Ni, Q.; Yao, Y.; Xu, H.; Zeng, B.; Li, Y.; Wei, Z. The revised complete mitogenome sequence of the tree frog Polypedates megacephalus (Anura, Rhacophoridae) by next-generation sequencing and phylogenetic analysis. PeerJ 2019, 7, e7415. [Google Scholar] [CrossRef]
- Fu, C.; Wang, Q.; Hu, T.; Lei, Z.; Fan, H.; Zhao, T.; Zong, H. The complete mitochondrial genome of Omei Treefrog (Rhacophorus omeimontis). Mitochondrial DNA Part B 2020, 5, 300–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, H.; Haramoto, Y.; Kubota, H.Y.; Shigeri, Y. Complete mitochondrial genome sequence of Japanese forest green tree frog (Rhacophorus arboreus). Mitochondrial DNA Part B 2020, 5, 3365–3366. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.R.; Wright, T.F.; Bermingham, E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol. Biol. Evol. 2001, 18, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Nie, L.; Wang, J.; Zhou, H.; Hou, H.; Wang, H.; Liu, J. Recombination and evolution of duplicate control regions in the mitochondrial genome of the Asian big-headed turtle, Platysternon megacephalum. PLoS ONE 2013, 8, e82854. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. Gene rearrangements in snake mitochondrial genomes: Highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol. Biol. Evol. 1996, 13, 1242–1254. [Google Scholar] [CrossRef]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Evolution of the deep-sea gulper eel mitochondrial genomes: Large-scale gene rearrangements originated within the eels. Mol. Biol. Evol. 2003, 20, 1917–1924. [Google Scholar] [CrossRef]
- Akiyama, T.; Nishida, C.; Momose, K.; Onuma, M.; Takami, K.; Masuda, R. Gene duplication and concerted evolution of mitochondrial DNA in crane species. Mol. Phylogenet. Evol. 2017, 106, 158–163. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, H.; Liang, D.; Liu, Y.-F.; Chen, Y.-Q.; Qu, L.-H. The complete mitochondrial genome of a tree frog, Polypedates megacephalus (Amphibia: Anura: Rhacophoridae), and a novel gene organization in living amphibians. Gene 2005, 346, 133–143. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Sano, N.; Kurabayashi, A.; Fujii, T.; Yonekawa, H.; Sumida, M. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of the bell-ring frog, Buergeria buergeri (family Rhacophoridae). Genes Genet. Syst. 2004, 79, 151–163. [Google Scholar] [CrossRef]
- Zhang, P.; Liang, D.; Mao, R.-L.; Hillis, D.M.; Wake, D.B.; Cannatella, D.C. Efficient sequencing of anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol. Biol. Evol. 2013, 30, 1899–1915. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Huang, M.; Lv, T.; Duan, R.; Zhang, S.; Li, H. The complete mitochondrial genome of Rhacophorus dennysi (Anura: Rhacophoridae) and phylogenetic analysis. Mitochondrial DNA Part A 2015, 27, 3719–3720. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Ranwez, V.; Douzery, E.J.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Bensch, S. Mitochondrial genomic rearrangements in songbirds. Mol. Biol. Evol. 2000, 17, 107–113. [Google Scholar] [CrossRef]
- Ogoh, K.; Ohmiya, Y. Concerted evolution of duplicated control regions within an ostracod mitochondrial genome. Mol. Biol. Evol. 2007, 24, 74–78. [Google Scholar] [CrossRef]
- Sumida, M.; Kanamori, Y.; Kaneda, H.; Kato, Y.; Nishioka, M.; Hasegawa, M.; Yonekawa, H. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of the Japanese pond frog Rana nigromaculata. Genes Genet. Syst. 2001, 76, 311–325. [Google Scholar] [CrossRef]
- AmphibiaWeb. University of California, Berkeley, CA, USA. Available online: https://amphibiaweb.org (accessed on 5 May 2021).
- Chen, W.-H.; Lu, G.; Bork, P.; Hu, S.; Lercher, M.J. Energy efficiency trade-offs drive nucleotide usage in transcribed regions. Nat. Commun. 2016, 7, 11334. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Y.; Yan, L.-P.; Pape, T.; Gao, Y.-Y.; Zhang, D. Evolutionary insights into bot flies (Insecta: Diptera: Oestridae) from comparative analysis of the mitochondrial genomes. Int. J. Biol. Macromol. 2020, 149, 371–380. [Google Scholar] [CrossRef]
- Yokobori, S.-I.; Fukuda, N.; Nakamura, M.; Aoyama, T.; Oshima, T. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol. Biol. Evol. 2004, 21, 2034–2046. [Google Scholar] [CrossRef]
- Yamazaki, N.; Ueshima, R.; Terrett, J.A.; Yokobori, S.-I.; Kaifu, M.; Segawa, R.; Kobayashi, T.; Numachi, K.-I.; Ueda, T.; Nishikawa, K. Evolution of pulmonate gastropod mitochondrial genomes: Comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics 1997, 145, 749–758. [Google Scholar] [CrossRef]
- Masta, S.E.; Boore, J.L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 2004, 21, 893–902. [Google Scholar] [CrossRef]
- Qiu, Y.; Song, D.; Zhou, K.; Sun, H. The mitochondrial sequences of Heptathela hangzhouensis and Ornithoctonus huwena reveal unique gene arrangements and atypical tRNAs. J. Mol. Evol. 2005, 60, 57–71. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Ma, Z.; Zhou, C. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Miya, M.; Kawaguchi, A.; Nishida, M. Mitogenomic exploration of higher teleostean phylogenies: A case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Biol. Evol. 2001, 18, 1993–2009. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, J.; Li, B.; Ouyang, B.; Yang, J. The complete mitochondrial genome of Budorcas taxicolor tibetana (Artiodactyla: Bovidae) and comparison with other Caprinae species: Insight into the phylogeny of the genus Budorcas. Int. J. Biol. Macromol. 2019, 121, 223–232. [Google Scholar] [CrossRef]
- Sun, C.-H.; Liu, H.-Y.; Min, X.; Lu, C.-H. Mitogenome of the little owl Athene noctua and phylogenetic analysis of Strigidae. Int. J. Biol. Macromol. 2020, 151, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Z.; Che, Y. The complete mitogenome of the wood-feeding cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and its phylogenetic relationship among cockroach Families. Int. J. Mol. Sci. 2017, 18, 2397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, Y.; Wu, Y.; Song, F.; Cai, W.; Li, H. Novel tRNA gene rearrangements in the mitochondrial genome of Camarochiloides weiweii (Hemiptera: Pachynomidae). Int. J. Biol. Macromol. 2020, 165, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, Y.; Wang, Y.; Liu, Z. Comparative analysis of five mitogenomes of Osmylinae (Neuroptera: Osmylidae) and their phylogenetic implications. Int. J. Biol. Macromol. 2020, 164, 447–455. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Steinberg, S.; Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1994, 1, 507–510. [Google Scholar] [CrossRef]
- Varani, G.; McClain, W.H. The G·U wobble base pair. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef]
- Wong, T.W.; Clayton, D.A. In vitro replication of human mitochondrial DNA: Accurate initiation at the origin of light-strand synthesis. Cell 1985, 42, 951–958. [Google Scholar] [CrossRef]
- Hixson, J.; Wong, T.; Clayton, D.A. Both the conserved stem-loop and divergent 5′-flanking sequences are required for initiation at the human mitochondrial origin of light-strand DNA replication. J. Biol. Chem. 1986, 261, 2384–2390. [Google Scholar] [CrossRef]
- Pereira, S.L. Mitochondrial genome organization and vertebrate phylogenetics. Genet. Mol. Biol. 2000, 23, 745–752. [Google Scholar] [CrossRef]
- Zardoya, R.; Garrido-Pertierra, A.; Bautista, J.M. The complete nucleotide sequence of the mitochondrial DNA genome of the rainbow trout, Oncorhynchus mykiss. J. Mol. Evol. 1995, 41, 942–951. [Google Scholar] [CrossRef]
- Ding, L.; Li, W.; Liao, J. Mitochondrial genome of Cricetulus migratorius (Rodentia: Cricetidae): Insights into the characteristics of the mitochondrial genome and the phylogenetic relationships of Cricetulus species. Gene 2016, 595, 121–129. [Google Scholar] [CrossRef]
- Desjardins, P.; Morais, R. Nucleotide sequence and evolution of coding and noncoding regions of a quail mitochondrial genome. J. Mol. Evol. 1991, 32, 153–161. [Google Scholar] [CrossRef]
- Haring, E.; Kruckenhauser, L.; Gamauf, A.; Riesing, M.J.; Pinsker, W. The complete sequence of the mitochondrial genome of Buteo buteo (Aves, Accipitridae) indicates an early split in the phylogeny of raptors. Mol. Biol. Evol. 2001, 18, 1892–1904. [Google Scholar] [CrossRef]
- Nishibori, M.; Tsudzuki, M.; Hayashi, T.; Yamamoto, Y.; Yasue, H. Complete nucleotide sequence of the Coturnix chinensis (blue-breasted quail) mitochondorial genome and a phylogenetic analysis with related species. J. Hered. 2002, 93, 439–444. [Google Scholar] [CrossRef]
- Igawa, T.; Kurabayashi, A.; Usuki, C.; Fujii, T.; Sumida, M. Complete mitochondrial genomes of three neobatrachian anurans: A case study of divergence time estimation using different data and calibration settings. Gene 2008, 407, 116–129. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Yoshikawa, N.; Sato, N.; Hayashi, Y.; Oumi, S.; Fujii, T.; Sumida, M. Complete mitochondrial DNA sequence of the endangered frog Odorrana ishikawae (family Ranidae) and unexpected diversity of mt gene arrangements in ranids. Mol. Phylogenet. Evol. 2010, 56, 543–553. [Google Scholar] [CrossRef]
- Kurabayashi, A.; Sumida, M. Afrobatrachian mitochondrial genomes: Genome reorganization, gene rearrangement mechanisms, and evolutionary trends of duplicated and rearranged genes. BMC Genom. 2013, 14, 633. [Google Scholar] [CrossRef]
- Jiang, D.; Jiang, K.; Ren, J.; Wu, J.; Li, J. Resurrection of the genus Leptomantis, with description of a new genus to the family Rhacophoridae (Amphibia: Anura). Asian Herpetol. Res. 2019, 10, 1–12. [Google Scholar]
- Madsen, C.S.; Ghivizzani, S.C.; Hauswirth, W.W. Protein binding to a single termination-associated sequence in the mitochondrial DNA D-loop region. Mol. Cell. Biol. 1993, 13, 2162–2171. [Google Scholar] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, H.; Robberson, D.L.; Vinograd, J. A novel closed-circular mitochondrial DNA with properties of a replicating intermediate. Proc. Natl. Acad. Sci. USA 1971, 68, 2252–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Mauro, D.; García-París, M.; Zardoya, R. Phylogenetic relationships of discoglossid frogs (Amphibia: Anura: Discoglossidae) based on complete mitochondrial genomes and nuclear genes. Gene 2004, 343, 357–366. [Google Scholar] [CrossRef]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. The complete nucleotide sequence of a snake (Dinodon semicarinatus) mitochondrial genome with two identical control regions. Genetics 1998, 150, 313–329. [Google Scholar] [CrossRef]
- Dowton, M.; Campbell, N.J. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Endo, K.; Noguchi, Y.; Ueshima, R.; Jacobs, H.T. Novel Repetitive Structures, Deviant Protein-Encoding Sequences andUnidentified ORFs in the Mitochondrial Genome of the Brachiopod Lingula anatina. J. Mol. Evol. 2005, 61, 36–53. [Google Scholar] [CrossRef]
- Mueller, R.L.; Boore, J.L. Molecular mechanisms of extensive mitochondrial gene rearrangement in plethodontid salamanders. Mol. Biol. Evol. 2005, 22, 2104–2112. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Recombination in animal mitochondrial DNA: Evidence from published sequences. Mol. Biol. Evol. 2001, 18, 2127–2131. [Google Scholar] [CrossRef]
- Abbott, C.L.; Double, M.C.; Trueman, J.W.; Robinson, A.; Cockburn, A. An unusual source of apparent mitochondrial heteroplasmy: Duplicate mitochondrial control regions in Thalassarche albatrosses. Mol. Ecol. 2005, 14, 3605–3613. [Google Scholar] [CrossRef]
- Verkuil, Y.I.; Piersma, T.; Baker, A.J. A novel mitochondrial gene order in shorebirds (Scolopacidae, Charadriiformes). Mol. Phylogenet. Evol. 2010, 57, 411–416. [Google Scholar] [CrossRef]
- Shao, R.; Barker, S.C.; Mitani, H.; Aoki, Y.; Fukunaga, M. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: A case study with Australasian Ixodes ticks. Mol. Biol. Evol. 2005, 22, 620–629. [Google Scholar] [CrossRef]
- Tatarenkov, A.; Avise, J.C. Rapid concerted evolution in animal mitochondrial DNA. Proc. R. Soc. B Biol. Sci. 2007, 274, 1795–1798. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.-L.; Nie, L.-W.; Pu, Y.-G. Complete mitochondrial genome of Chinese big-headed turtle, Platysternon megacephalum, with a novel gene organization in vertebrate mtDNA. Gene 2006, 380, 14–20. [Google Scholar] [CrossRef]
- Schirtzinger, E.E.; Tavares, E.S.; Gonzales, L.A.; Eberhard, J.R.; Miyaki, C.Y.; Sanchez, J.J.; Hernandez, A.; Müeller, H.; Graves, G.R.; Fleischer, R.C. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol. Phylogenet. Evol. 2012, 64, 342–356. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Wright, T.F. Rearrangement and evolution of mitochondrial genomes in parrots. Mol. Phylogenet. Evol. 2016, 94, 34–46. [Google Scholar] [CrossRef]
- Sloan, D.B.; Müller, K.; McCauley, D.E.; Taylor, D.R.; Štorchová, H. Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 2012, 196, 1228–1239. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; Zhang, Q.; Chen, L.; Shu, F.; Ma, X.; Bian, Y. The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 2020, 143, 373–381. [Google Scholar] [CrossRef]
- Wang, X.; Song, A.; Wang, F.; Chen, M.; Li, X.; Li, Q.; Liu, N. The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Int. J. Biol. Macromol. 2020, 162, 209–219. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M.; Harrison, R.G. Mitochondrial DNA transmission genetics in crickets. Genetics 1986, 114, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Zouros, E.; Ball, A.O.; Saavedra, C.; Freeman, K.R. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc. Natl. Acad. Sci. USA 1994, 91, 7463–7467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Cheng, C.-H.C. ND6 gene “lost” and found: Evolution of mitochondrial gene rearrangement in Antarctic notothenioids. Mol. Biol. Evol. 2010, 27, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.J.; Castoe, T.A.; Austin, C.C.; Burbrink, F.T.; Herron, M.D.; McGuire, J.A.; Parkinson, C.L.; Pollock, D.D. Comparative mitochondrial genomics of snakes: Extraordinary substitution rate dynamics and functionality of the duplicate control region. BMC Evol. Biol. 2007, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Black, W., 4th; Roehrdanz, R.L. Mitochondrial gene order is not conserved in arthropods: Prostriate and metastriate tick mitochondrial genomes. Mol. Biol. Evol. 1998, 15, 1772–1785. [Google Scholar] [CrossRef]
- Mindell, D.P.; Sorenson, M.D.; Dimcheff, D.E. Multiple independent origins of mitochondrial gene order in birds. Proc. Natl. Acad. Sci. USA 1998, 95, 10693–10697. [Google Scholar] [CrossRef]
- Scouras, A.; Beckenbach, K.; Arndt, A.; Smith, M.J. Complete mitochondrial genome DNA sequence for two ophiuroids and a holothuroid: The utility of protein gene sequence and gene maps in the analyses of deep deuterostome phylogeny. Mol. Phylogenet. Evol. 2004, 31, 50–65. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Hewitt, G.M. Highly conserved nuclear copies of the mitochondrial control region in the desert locust Schistocerca gregaria: Some implications for population studies. Mol. Ecol. 1996, 5, 295–300. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics 2001, 157, 621–637. [Google Scholar] [CrossRef]
- Uliano-Silva, M.; Americo, J.A.; Costa, I.; Schomaker-Bastos, A.; de Freitas Rebelo, M.; Prosdocimi, F. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of Mytilidae. Gene 2016, 577, 202–208. [Google Scholar] [CrossRef]
- Steinauer, M.L.; Nickol, B.B.; Broughton, R.; Ortí, G. First sequenced mitochondrial genome from the phylum Acanthocephala (Leptorhynchoides thecatus) and its phylogenetic position within Metazoa. J. Mol. Evol. 2005, 60, 706–715. [Google Scholar] [CrossRef]
- Suga, K.; Mark Welch, D.B.; Tanaka, Y.; Sakakura, Y.; Hagiwara, A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 2008, 25, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Pyron, R.A.; Wiens, J.J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogenet. Evol. 2011, 61, 543–583. [Google Scholar] [CrossRef]
- Irisarri, I.; San Mauro, D.; Abascal, F.; Ohler, A.; Vences, M.; Zardoya, R. The origin of modern frogs (Neobatrachia) was accompanied by acceleration in mitochondrial and nuclear substitution rates. BMC Genom. 2012, 13, 626. [Google Scholar] [CrossRef]
- Chen, G.; Wang, B.; Liu, J.; Xie, F.; Jiang, J. Complete mitochondrial genome of Nanorana pleskei (Amphibia: Anura: Dicroglossidae) and evolutionary characteristics. Curr. Zool. 2011, 57, 785–805. [Google Scholar] [CrossRef]
- Li, J.-T.; Li, Y.; Klaus, S.; Rao, D.-Q.; Hillis, D.M.; Zhang, Y.-P. Diversification of rhacophorid frogs provides evidence for accelerated faunal exchange between India and Eurasia during the Oligocene. Proc. Natl. Acad. Sci. USA 2013, 110, 3441–3446. [Google Scholar] [CrossRef]
- Yuan, Z.-Y.; Zhang, B.-L.; Raxworthy, C.J.; Weisrock, D.W.; Hime, P.M.; Jin, J.-Q.; Lemmon, E.M.; Lemmon, A.R.; Holland, S.D.; Kortyna, M.L. Natatanuran frogs used the Indian Plate to step-stone disperse and radiate across the Indian Ocean. Natl. Sci. Rev. 2018, 6, 10–14. [Google Scholar] [CrossRef]
- Ren, Z.; Zhu, B.; Ma, E.; Wen, J.; Tu, T.; Cao, Y.; Hasegawa, M.; Zhong, Y. Complete nucleotide sequence and gene arrangement of the mitochondrial genome of the crab-eating frog Fejervarya cancrivora and evolutionary implications. Gene 2009, 441, 148–155. [Google Scholar] [CrossRef]
- Li, J.-N.; Liang, D.; Wang, Y.-Y.; Guo, P.; Huang, S.; Zhang, P. A large-scale systematic framework of Chinese snakes based on a unified multilocus marker system. Mol. Phylogenet. Evol. 2020, 148, 106807. [Google Scholar] [CrossRef]
- Macey, J.R.; Schulte, J.A.; Larson, A. Evolution and phylogenetic information content of mitochondrial genomic structural features illustrated with acrodont lizards. Syst. Biol. 2000, 49, 257–277. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Fragments | Primer Names | Primer Nucleotide Sequences (5′-3′) | Length of Fragments |
|---|---|---|---|
| F1 | COIIF a | GACTCACTCAAGCGTCTATTC | ~1300 bp |
| ATP6R a | TGTGGGCGGGTTTATT | ||
| F2 | CytbFow1 b | GTYCTMCCNTGRGGHCAAATATCHTTYTG | ~3500 bp |
| CytbF2 a | TTAGCCCTTCTATCTACCCTC | ||
| FND512800H c | CCTATTTTDCGRATRTCYTGYTC | ||
| ND5R3 a | CAGCCAATAAGTAAATAGGACA | ||
| F3 | ND5F2 a | CTCACCCCTCTATTACGACTT | ~5000 bp |
| ND5Fow_sch b | TGACTWGCMGCAGCAATAGAAGG | ||
| ND5F3 a | CCCGCTGTTATGACTTGGAA | ||
| R16M1 d | GGGTATCTAATCCCAGTTTG | ||
| R17N1 b | GCTGAGACTTGCATGTGTAA | ||
| 12SR1 a | TTAACTTGAGTCCGCCGGTA | ||
| 12S600H c | TCGATTATAGAACAGGCTCCTCT |
| Region | AT Content (%) | GC Content (%) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | A | B | C | D | E | F | G | H | I | ||
| Genome | 61.0 | 60.9 | 60.6 | 61.6 | 62.1 | 62.4 | 63.0 | 64.5 | 60.4 | 39.0 | 39.1 | 39.4 | 38.4 | 38.0 | 37.5 | 37.0 | 35.5 | 39.5 | |
| All PCGs | 59.9 | 59.3 | 59.2 | 59.9 | 59.4 | 61.7 | 62.3 | 60.9 | 59.0 | 40.2 | 40.6 | 40.9 | 40.1 | 40.6 | 38.2 | 37.7 | 39.2 | 41.0 | |
| 1st Codon | 60.8 | 60.1 | 58.6 | 59.3 | 53.9 | 54.9 | 56.1 | 55.2 | 52.7 | 39.2 | 39.9 | 41.5 | 40.7 | 46.1 | 45.1 | 43.9 | 44.8 | 47.4 | |
| 2nd Codon | 57.5 | 57.2 | 59.6 | 60.5 | 59.6 | 59.5 | 60.5 | 59.6 | 59.6 | 42.5 | 42.8 | 40.5 | 39.5 | 40.4 | 40.5 | 39.4 | 40.5 | 40.4 | |
| 3rd Codon | 61.1 | 60.7 | 59.3 | 59.8 | 64.7 | 70.8 | 70.4 | 67.8 | 64.6 | 38.8 | 39.3 | 40.7 | 40.3 | 35.3 | 29.2 | 29.6 | 32.2 | 35.3 | |
| All tRNAs | 57.5 | 57.3 | 57.4 | 57.9 | 58.9 | 59.8 | 59.5 | 58.2 | 56.3 | 42.4 | 42.6 | 42.6 | 42.2 | 41.2 | 40.2 | 40.5 | 41.8 | 43.6 | |
| All rRNAs | 59.0 | 59.9 | 59.2 | 59.7 | 59.5 | 60.7 | 60.8 | 59.8 | 58.0 | 41.0 | 40.0 | 40.7 | 40.3 | 40.6 | 39.3 | 39.2 | 40.2 | 42.0 | |
| CR1 | 64.3 | 65.4 | 65.7 | 65.9 | 68.4 | 68.8 | 64.4 | 65.2 | 66.8 | 35.7 | 34.6 | 34.3 | 34.1 | 31.6 | 31.2 | 35.6 | 34.8 | 33.2 | |
| CR2 | 65.0 | 67.0 | 68.8 | 69.7 | 69.3 | N/A | 67.6 | 69.8 | N/A | 35.0 | 32.9 | 31.3 | 30.2 | 30.7 | N/A | 32.4 | 30.2 | N/A | |
| Region | AT Skew | GC Skew | |||||||||||||||||
| A | B | C | D | E | F | G | H | I | A | B | C | D | E | F | G | H | I | ||
| Genome | −0.010 | 0.002 | −0.003 | −0.036 | 0.024 | 0.006 | 0.032 | 0.020 | −0.010 | −0.246 | −0.258 | −0.249 | −0.214 | −0.226 | −0.237 | −0.259 | −0.251 | −0.256 | |
| All PCGs | −0.085 | −0.073 | −0.078 | −0.105 | −0.044 | −0.057 | −0.059 | −0.048 | −0.078 | −0.239 | −0.241 | −0.247 | −0.197 | −0.281 | −0.257 | −0.268 | −0.286 | −0.278 | |
| 1st Codon | 0.007 | 0.042 | −0.085 | −0.093 | 0.083 | 0.064 | 0.084 | 0.072 | 0.089 | −0.224 | −0.233 | −0.210 | −0.174 | 0.015 | 0.033 | 0.021 | 0.013 | 0.017 | |
| 2nd Codon | −0.085 | −0.094 | −0.060 | −0.107 | −0.386 | −0.382 | −0.392 | −0.386 | −0.379 | −0.186 | −0.173 | −0.284 | −0.235 | −0.371 | −0.363 | −0.371 | −0.373 | −0.386 | |
| 3rd Codon | −0.175 | −0.166 | −0.086 | −0.117 | 0.172 | 0.124 | 0.114 | 0.150 | 0.062 | −0.309 | −0.328 | −0.248 | −0.186 | −0.564 | −0.555 | −0.561 | −0.596 | −0.547 | |
| All tRNAs | 0.026 | 0.026 | 0.031 | 0.022 | 0.032 | 0.030 | 0.025 | 0.024 | 0.030 | 0.033 | 0.028 | 0.033 | 0.057 | 0.019 | 0.045 | 0.037 | 0.048 | 0.005 | |
| All rRNAs | 0.146 | 0.145 | 0.146 | 0.128 | 0.181 | 0.174 | 0.191 | 0.188 | 0.146 | −0.090 | −0.072 | −0.101 | −0.076 | −0.094 | −0.090 | −0.096 | −0.106 | −0.109 | |
| CR1 | 0.020 | 0.021 | 0.014 | −0.017 | −0.066 | 0.000 | 0.121 | 0.086 | −0.010 | −0.333 | −0.353 | −0.294 | −0.308 | 0.032 | −0.200 | −0.234 | −0.206 | −0.184 | |
| CR2 | −0.006 | 0.000 | −0.033 | −0.076 | 0.065 | N/A | 0.071 | 0.094 | N/A | −0.326 | −0.343 | −0.252 | −0.199 | −0.173 | N/A | −0.255 | −0.265 | N/A | |
| Anurans with Duplicated CRs | GenBank ID | Length of CR1 | Length of CR2 | Length of Similar Regions/Similarity |
|---|---|---|---|---|
| Breviceps adspersus | NC_023379.1 | 6466 bp | 4018 bp | 3148 bp/99.0% |
| Hoplobatrachus rugulosus | NC_019615.1 | 1815 bp | 1772 bp | 1772 bp/97.2% |
| Hoplobatrachus tigerinus | NC_014581.1 | 3415 bp | 1586 bp | 1586 bp/99.2% |
| Hyperolius marmoratus | NC_023381.1 | 2014 bp | 2286 bp | 1857 bp/99.6% |
| Mantella baroni | NC_039758.1 | 2168 bp | 2970 bp | 2099 bp/94.7% |
| Mantella madagascariensis | NC_007888.1 | 4704 bp | 2274 bp | 2217 bp/93.7% |
| Polypedates braueri | NC_042797.1 | 1753 bp | 2797 bp | 1524 bp/99.9% |
| Polypedates impresus | MN869008.1 | 1401 bp | 2264 bp | 1496 bp/91.1% |
| Polypedates megacephalus | NC_043955.1 | 1574 bp | 2230 bp | 1571 bp/99.5% |
| Polypedates mutus | MN869009.1 | 1959 bp | 2760 bp | 1690 bp/99.7% |
| Rana amurensis | NC_030042.1 | 2695 bp | 2324 bp | 1708 bp/94.2% |
| Rana kunyuesis | NC_024548.1 | 3969 bp | 2777 bp | 2366 bp/96.8% |
| Trichobatrachus robustus | NC_023382.1 | 1699 bp | 1788 bp | 1390 bp/99.7% |
| Zhangixalus arboreus | LC565708.1 | 3617 bp | 3345 bp | 1718 bp/99.4% |
| Zhangixalus omeimontis | NC_046387.1 | 1822 bp | 2214 bp | 1506 bp/99.0% |
| Zhangixalus schlegelii | NC_007178.1 | 2688 bp | 3272 bp | 1510 bp/97.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, L.; Huang, A.; He, Z.; Ao, L.; Ge, F.; Fan, X.; Zeng, B.; Yang, M.; Yang, D.; Ni, Q.; et al. Complete Mitogenomes of Polypedates Tree Frogs Unveil Gene Rearrangement and Concerted Evolution within Rhacophoridae. Animals 2022, 12, 2449. https://doi.org/10.3390/ani12182449
Cui L, Huang A, He Z, Ao L, Ge F, Fan X, Zeng B, Yang M, Yang D, Ni Q, et al. Complete Mitogenomes of Polypedates Tree Frogs Unveil Gene Rearrangement and Concerted Evolution within Rhacophoridae. Animals. 2022; 12(18):2449. https://doi.org/10.3390/ani12182449
Chicago/Turabian StyleCui, Lin, An Huang, Zhi He, Lisha Ao, Fei Ge, Xiaolan Fan, Bo Zeng, Mingyao Yang, Deying Yang, Qingyong Ni, and et al. 2022. "Complete Mitogenomes of Polypedates Tree Frogs Unveil Gene Rearrangement and Concerted Evolution within Rhacophoridae" Animals 12, no. 18: 2449. https://doi.org/10.3390/ani12182449