
The Effects of Breed and Residual Feed Intake Divergence on the Abundance and Active Population of Rumen Microbiota in Beef Cattle


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments and Sample Collection
2.2. Nucleic Acid Extractions and cDNA Synthesis
2.3. Quantitative Real Time PCR Analysis
2.4. Statistical Analysis
3. Results
3.1. Assessment of Four Targeted Microbial Groups in Rumen of Beef Steers
3.2. Effects of Breed and RFI on the Abundance of the Four Microbial Groups in the Rumen
3.3. Effects of Breed and RFI on Active Populations of the Four Microbial Groups in the Rumen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kenny, D.A.; Fitzsimons, C.; Waters, S.M.; McGee, M. Invited review: Improving feed efficiency of beef cattle–the current state of the art and future challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savietto, D.; Berry, D.P.; Friggens, N.C. Towards an improved estimation of the biological components of residual feed intake in growing cattle. J. Anim. Sci. 2014, 92, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.P.; Crowley, J.J. Cell biology symposium: Genetics of feed efficiency in dairy and beef cattle. J. Anim. Sci. 2013, 91, 1594–1613. [Google Scholar] [CrossRef] [Green Version]
- Herd, R.M.; Arthur, P.F. Physiological basis for residual feed intake. J. Anim. Sci. 2009, 87, E64–E71. [Google Scholar] [CrossRef] [PubMed]
- Montanholi, Y.; Fontoura, A.; Swanson, K.; Coomber, B.; Yamashiro, S.; Miller, S. Small intestine histomorphometry of beef cattle with divergent feed efficiency. Acta Vet. Scand. 2013, 55, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, L.L.; Nkrumah, J.D.; Basarab, J.A.; Moore, S.S. Linkage of microbial ecology to phenotype: Correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol. Lett. 2008, 288, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Sanabria, E.; Guan, L.L.; Goonewardene, L.A.; Li, M.; Mujibi, D.F.; Stothard, P.; Moore, S.S.; Leon-Quintero, M.C. Correlation of Particular Bacterial PCR-Denaturing Gradient Gel Electrophoresis Patterns with Bovine Ruminal Fermentation Parameters and Feed Efficiency Traits. Appl. Environ. Microb. 2010, 76, 6338–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myer, P.R.; Smith, T.P.L.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C. Rumen Microbiome from Steers Differing in Feed Efficiency. PLoS ONE 2015, 10, e129174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabat, S.K.B.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Assessment of the Microbial Ecology of Ruminal Methanogens in Cattle with Different Feed Efficiencies. Appl. Environ. Microb. 2009, 75, 6524–6533. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Characterization of Variation in Rumen Methanogenic Communities under Different Dietary and Host Feed Efficiency Conditions, as Determined by PCR-Denaturing Gradient Gel Electrophoresis Analysis. Appl. Environ. Microb. 2010, 76, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carberry, C.A.; Waters, S.M.; Kenny, D.A.; Creevey, C.J. Rumen Methanogenic Genotypes Differ in Abundance According to Host Residual Feed Intake Phenotype and Diet Type. Appl. Environ. Microb. 2014, 80, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Guan, L.L. Metatranscriptomic Profiling Reveals Linkages between the Active Rumen Microbiome and Feed Efficiency in Beef Cattle. Appl. Environ. Microb. 2017, 83, e00061-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Hitch, T.C.A.; Chen, Y.; Creevey, C.J.; Guan, L.L. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Henderson, G.; Sun, X.; Cox, F.; Janssen, P.H.; Guan, L.L. Taxonomic Assessment of Rumen Microbiota Using Total RNA and Targeted Amplicon Sequencing Approaches. Front. Microbiol. 2016, 7, 987. [Google Scholar] [CrossRef] [Green Version]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [Green Version]
- Puniya, A.K.; Kamra, D.N.; Singh, R. Rumen Microbiology: From Evolution to Revolution; Springer: New Delhi, India, 2015; p. 380. [Google Scholar]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Sanabria, E.; Goonewardene, L.A.; Wang, Z.; Zhou, M.; Moore, S.S.; Guan, L.L. Influence of Sire Breed on the Interplay among Rumen Microbial Populations Inhabiting the Rumen Liquid of the Progeny in Beef Cattle. PLoS ONE 2013, 8, e58461. [Google Scholar] [CrossRef] [Green Version]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Forano, E.; Martin, C.; Newbold, C.J. Microbial ecosystem and methanogenesis in ruminants. Animal 2010, 4, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, D.J. Genetics of efficient feed utilization and national cattle evaluation: A review. Genet. Mol. Res. 2005, 4, 152–165. [Google Scholar] [PubMed]
- Nkrumah, J.D.; Crews, D.H.J.; Basarab, J.A.; Price, M.A.; Okine, E.K.; Wang, Z.; Li, C.; Moore, S.S. Genetic and phenotypic relationships of feeding behavior and temperament with performance, feed efficiency, ultrasound, and carcass merit of beef cattle. J. Anim. Sci. 2007, 85, 2382–2390. [Google Scholar] [CrossRef]
- Olfert, D.E.D.; Cross, D.B.M.; McWilliam, M.A.A. Guide to the Care and Use of Experimental Animals; Canadian Council on Animal Care: Ottawa, ON, Canada, 1993; pp. 10–160. [Google Scholar]
- Nkrumah, J.D.; Okine, E.K.; Mathison, G.W.; Schmid, K.; Li, C.; Basarab, J.A.; Price, M.A.; Wang, Z.; Moore, S.S. Relationships of feedlot feed efficiency, performance, and feeding behavior with metabolic rate, methane production, and energy partitioning in beef cattle1. J. Anim. Sci. 2006, 84, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biot. 2007, 75, 165–174. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Karnati, S.K.R.; Yu, Z.; Morrison, M.; Firkins, J.L. Development of an Assay to Quantify Rumen Ciliate Protozoal Biomass in Cows Using Real-Time PCR. J. Nutr. 2004, 134, 3378–3384. [Google Scholar] [CrossRef] [Green Version]
- Kittelmann, S.; Naylor, G.E.; Koolaard, J.P.; Janssen, P.H. A proposed taxonomy of anaerobic fungi (class Neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS ONE 2012, 7, e36866. [Google Scholar] [CrossRef]
- Sirohi, S.K.; Singh, N.; Dagar, S.S.; Puniya, A.K. Molecular tools for deciphering the microbial community structure and diversity in rumen ecosystem. Appl. Microbiol. Biot. 2012, 95, 1135–1154. [Google Scholar] [CrossRef]
- Gaidos, E.; Rusch, A.; Ilardo, M. Ribosomal tag pyrosequencing of DNA and RNA from benthic coral reef microbiota: Community spatial structure, rare members and nitrogen-cycling guilds. Environ. Microbiol. 2011, 13, 1138–1152. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Marcelino, L.A.; Klepac-Ceraj, V.; Polz, M.F. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 2004, 186, 2629–2635. [Google Scholar] [CrossRef] [Green Version]
- Kembel, S.W.; Wu, M.; Eisen, J.A.; Green, J.L. Incorporating 16S Gene Copy Number Information Improves Estimates of Microbial Diversity and Abundance. PLoS Comput. Biol. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Angly, F.; Dennis, P.; Skarshewski, A.; Vanwonterghem, I.; Hugenholtz, P.; Tyson, G. CopyRighter: A rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome 2014, 2, 11. [Google Scholar] [CrossRef]
- Gong, J.; Dong, J.; Liu, X.; Massana, R. Extremely High Copy Numbers and Polymorphisms of the rDNA Operon Estimated from Single Cell Analysis of Oligotrich and Peritrich Ciliates. Protist 2013, 164, 369–379. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Wang, Y.; Katz, L.A.; Gao, F.; Song, W. Disentangling sources of variation in SSU rDNA sequences from single cell analyses of ciliates: Impact of copy number variation and experimental error. Proceedings. Biol. Sci. 2017, 284, 20170425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.G.; Klieve, A.V. Does the complexity of the rumen microbial ecology preclude methane mitigation? Anim. Feed Sci. Tech. 2011, 166–167, 248–253. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Wallace, R.J.; Rooke, J.A.; Duthie, C.; Hyslop, J.J.; Ross, D.W.; McKain, N.; de Souza, S.M.; Snelling, T.J.; Waterhouse, A.; Roehe, R. Archaeal abundance in post-mortem ruminal digesta may help predict methane emissions from beef cattle. Sci. Rep. 2014, 4, srep05892. [Google Scholar] [CrossRef]
- Tapio, I.; Snelling, T.J.; Strozzi, F.; Wallace, R.J. The ruminal microbiome associated with methane emissions from ruminant livestock. J. Anim. Sci. Biotechno. 2017, 8, 7. [Google Scholar] [CrossRef]
- Paz, H.A.; Anderson, C.L.; Muller, M.J.; Kononoff, P.J.; Fernando, S.C. Rumen Bacterial Community Composition in Holstein and Jersey Cows Is Different under Same Dietary Condition and Is Not Affected by Sampling Method. Front. Microbiol. 2016, 7, 1206. [Google Scholar] [CrossRef] [PubMed]
- Kittelmann, S.; Janssen, P.H. Characterization of rumen ciliate community composition in domestic sheep, deer, and cattle, feeding on varying diets, by means of PCR-DGGE and clone libraries. FEMS Microbiol. Ecol. 2011, 75, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Sanabria, E.; Goonewardene, L.A.; Wang, Z.; Durunna, O.N.; Moore, S.S. Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl. Environ. Microbiol. 2011, 78, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, P.; Kenny, D.A.; Earley, B.; McGee, M. Grazed grass herbage intake and performance of beef heifers with predetermined phenotypic residual feed intake classification. Animal 2012, 6, 1648–1661. [Google Scholar] [CrossRef] [Green Version]
- McCann, J.C.; Wiley, L.M.; Forbes, T.D.; Rouquette, J.F.M.; Tedeschi, L.O. Relationship between the rumen microbiome and residual feed intake-efficiency of Brahman bulls stocked on bermudagrass pastures. PLoS ONE 2014, 9, e91864. [Google Scholar] [CrossRef] [Green Version]


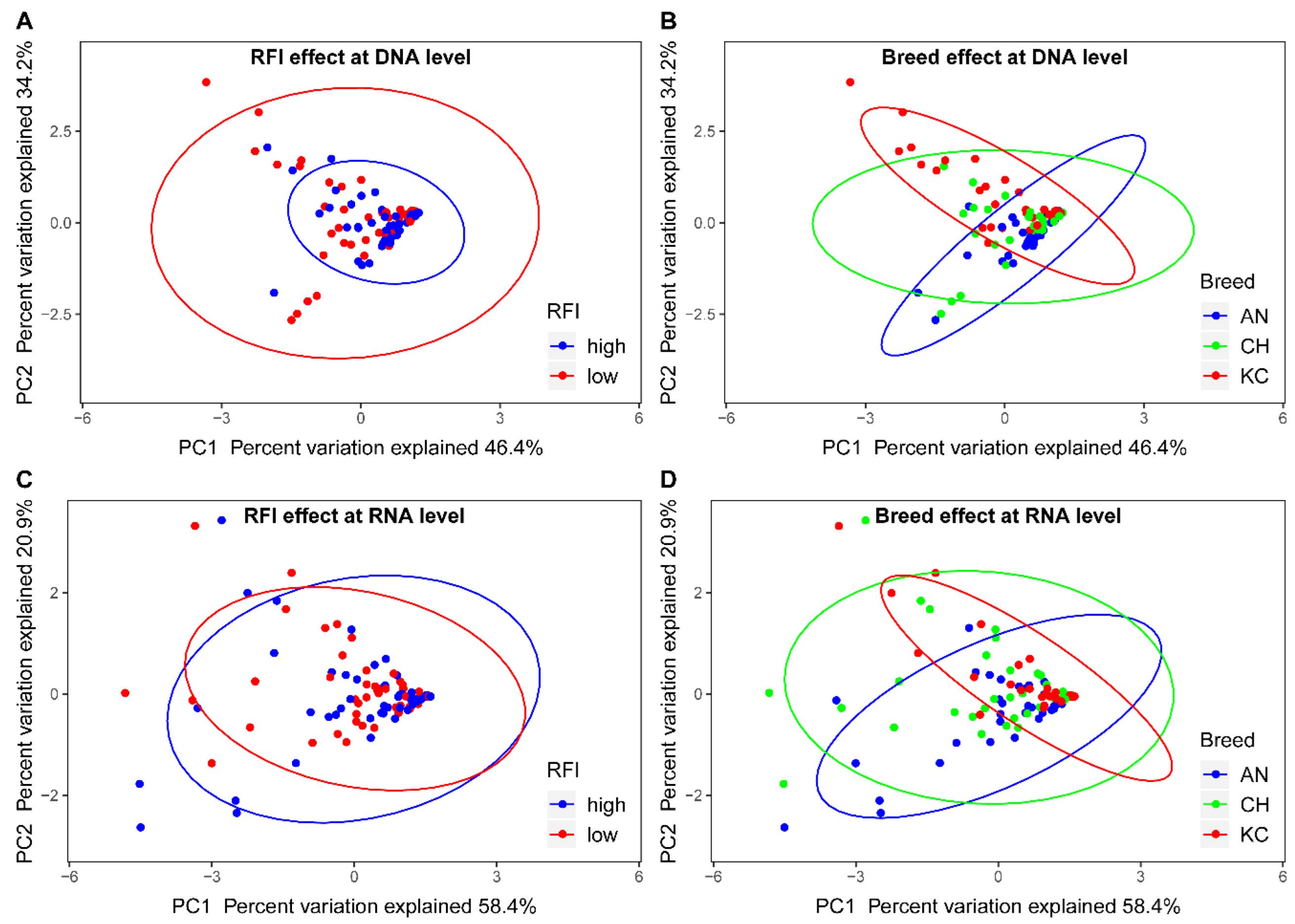{kind=link}
| Item | DNA | RNA | RNA-DNA | Fold Change (RNA/DNA) | p Value |
|---|---|---|---|---|---|
| Bacteria | 11.30 ± 0.36 a | 10.68 ± 0.36 b | −0.62 ± 0.11 | 1/4.17 | <0.01 |
| Archaea | 9.56 ± 0.24 a | 9.19 ± 0.24 b | −0.37 ± 0.10 | 1/2.34 | <0.01 |
| Protozoa | 8.70 ± 0.27 b | 10.46 ± 0.27 a | 1.77 ± 0.15 | 58.88 | <0.01 |
| Fungi | 5.47 ± 0.51 a | 4.32 ± 0.51 b | −1.14 ± 0.10 | 1/13.80 | <0.01 |
| Item | Mean | SD. | Bacteria | Archaea | Protozoa | Fungi |
|---|---|---|---|---|---|---|
| DNA | ||||||
| Bacteria | 11.30 | 0.41 | 1 | |||
| Archaea | 9.56 | 0.37 | 0.57 a | 1 | ||
| Protozoa | 8.70 | 1.03 | 0.44 a | 0.23 b | 1 | |
| Fungi | 5.47 | 1.40 | 0.17 c | 0.26 b | 0.54 a | 1 |
| RNA | ||||||
| Bacteria | 10.68 | 1.11 | 1 | |||
| Archaea | 9.19 | 1.04 | 0.93 a | 1 | ||
| Protozoa | 10.46 | 1.40 | 0.68 a | 0.81 a | 1 | |
| Fungi | 4.32 | 1.06 | 0.35 a | 0.30 a | 0.43 a | 1 |
| Items | Breed | RFI | p Value | |||||
|---|---|---|---|---|---|---|---|---|
| Angus | Charolais | Kinsella | High | Low | Breed | RFI | Breed × RFI | |
| Bacteria | 11.41 ± 0.12 a | 11.24 ± 0.18 ab | 11.10 ± 0.08 b | 11.22 ± 0.12 | 11.28 ± 0.11 | <0.01 | 0.53 | 0.23 |
| Archaea | 9.55 ± 0.12 | 9.52 ± 0.18 | 9.44 ± 0.07 | 9.46 ± 0.12 | 9.55 ± 0.11 | 0.60 | 0.20 | 0.47 |
| Protozoa | 8.24 ± 0.30 b | 8.86 ± 0.45 a | 9.08 ± 0.19 a | 8.62 ± 0.30 | 8.82 ± 0.29 | <0.01 | 0.27 | 0.53 |
| Fungi | 5.06 ± 0.31 b | 5.26 ± 0.42 b | 6.32 ± 0.24 a | 5.36 ± 0.29 | 5.75 ± 0.28 | <0.01 | 0.13 | 0.80 |
| Item | Breeds | RFI | p Value | |||||
|---|---|---|---|---|---|---|---|---|
| Angus | Charolais | Kinsella | High | Low | Breeds | RFI | Breeds × RFI | |
| Bacteria | 11.12 ± 0.27 a | 10.87 ± 0.40 a | 9.76 ± 0.19 b | 10.60 ± 0.26 | 10.57 ± 0.26 | <0.01 | 0.86 | 0.92 |
| Archaea | 9.58 ± 0.16 a | 9.62 ± 0.16 a | 8.36 ± 0.16 b | 9.20 ± 0.13 | 9.18 ± 0.13 | <0.01 | 0.91 | 0.95 |
| Protozoa | 10.38 ± 0.29 a | 10.87 ± 0.36 a | 10.02 ± 0.25 b | 10.39 ± 0.26 | 10.47 ± 0.26 | <0.10 | 0.78 | 0.77 |
| Fungi | 4.35 ± 0.35 | 4.38 ± 0.54 | 4.62 ± 0.21 | 4.40 ± 0.35 | 4.50 ± 0.34 | 0.65 | 0.61 | 0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Li, F.; Chen, Y.; Guan, L.-L. The Effects of Breed and Residual Feed Intake Divergence on the Abundance and Active Population of Rumen Microbiota in Beef Cattle. Animals 2022, 12, 1966. https://doi.org/10.3390/ani12151966
Zhang Y, Li F, Chen Y, Guan L-L. The Effects of Breed and Residual Feed Intake Divergence on the Abundance and Active Population of Rumen Microbiota in Beef Cattle. Animals. 2022; 12(15):1966. https://doi.org/10.3390/ani12151966
Chicago/Turabian StyleZhang, Yawei, Fuyong Li, Yanhong Chen, and Le-Luo Guan. 2022. "The Effects of Breed and Residual Feed Intake Divergence on the Abundance and Active Population of Rumen Microbiota in Beef Cattle" Animals 12, no. 15: 1966. https://doi.org/10.3390/ani12151966