
Genotype Shift of Malaysian Porcine Circovirus 2 (PCV2) from PCV2b to PCV2d within a Decade

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Molecular Detection of PCV2
2.3. Pairwise Sequence Comparison (PASC) Analysis
2.4. Phylogenetic Analysis and Genotyping
2.5. Nucleotide Sequence Polymorphism Analysis
2.6. Rates of Substitution
3. Results
3.1. Molecular Detection of PCV2
3.2. Pairwise Sequence Comparison (PASC)
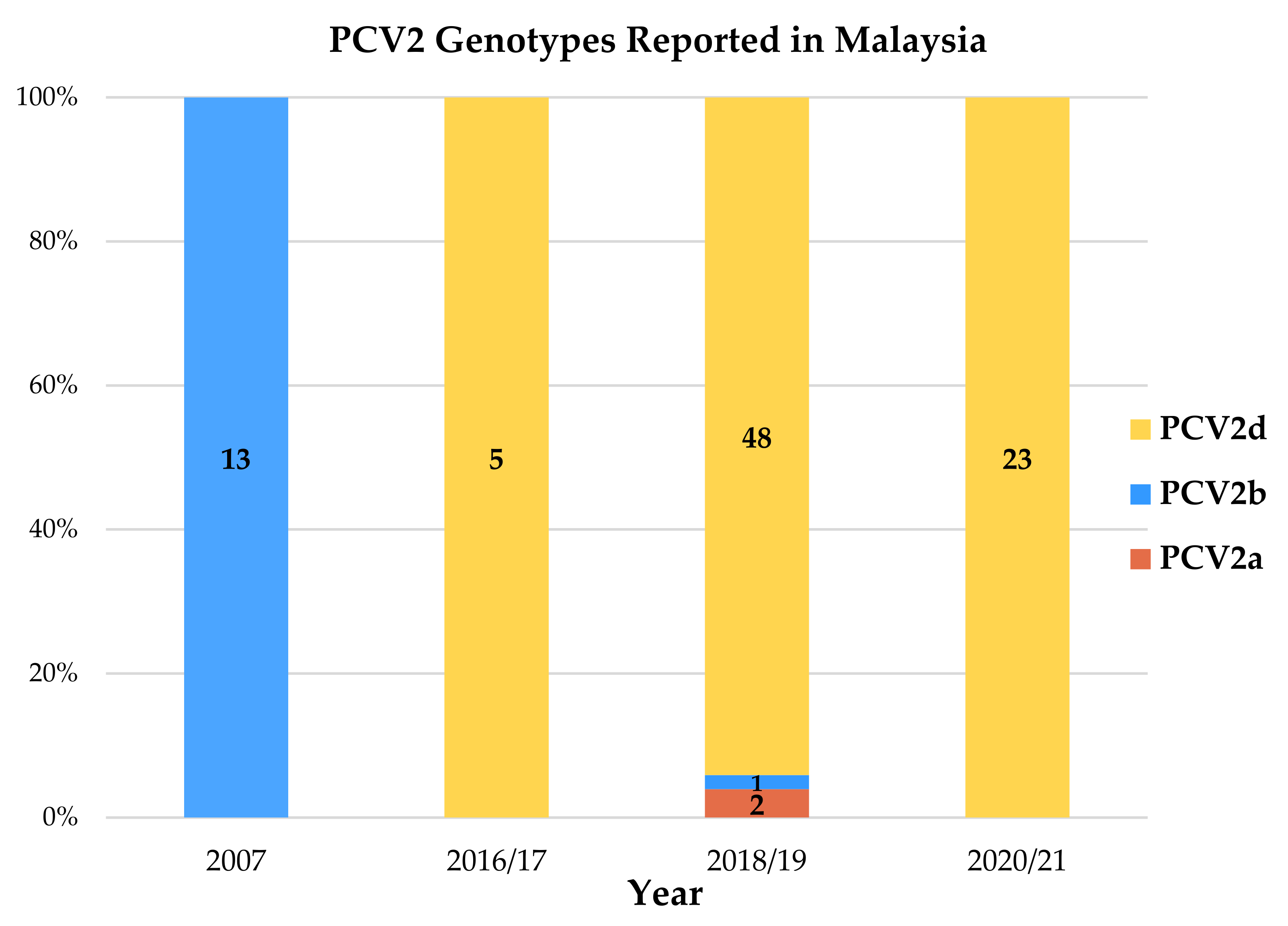
3.3. Phylogenetic Analysis and Genotyping
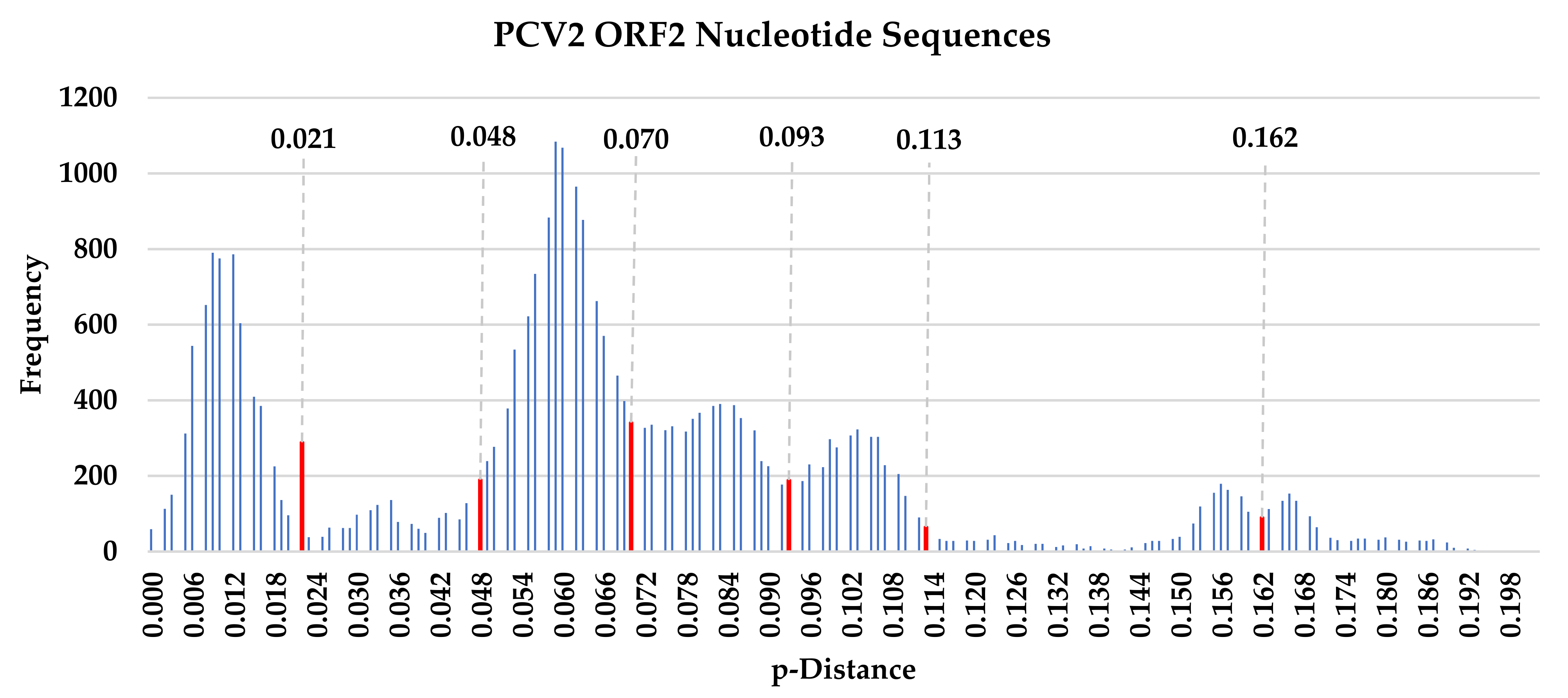
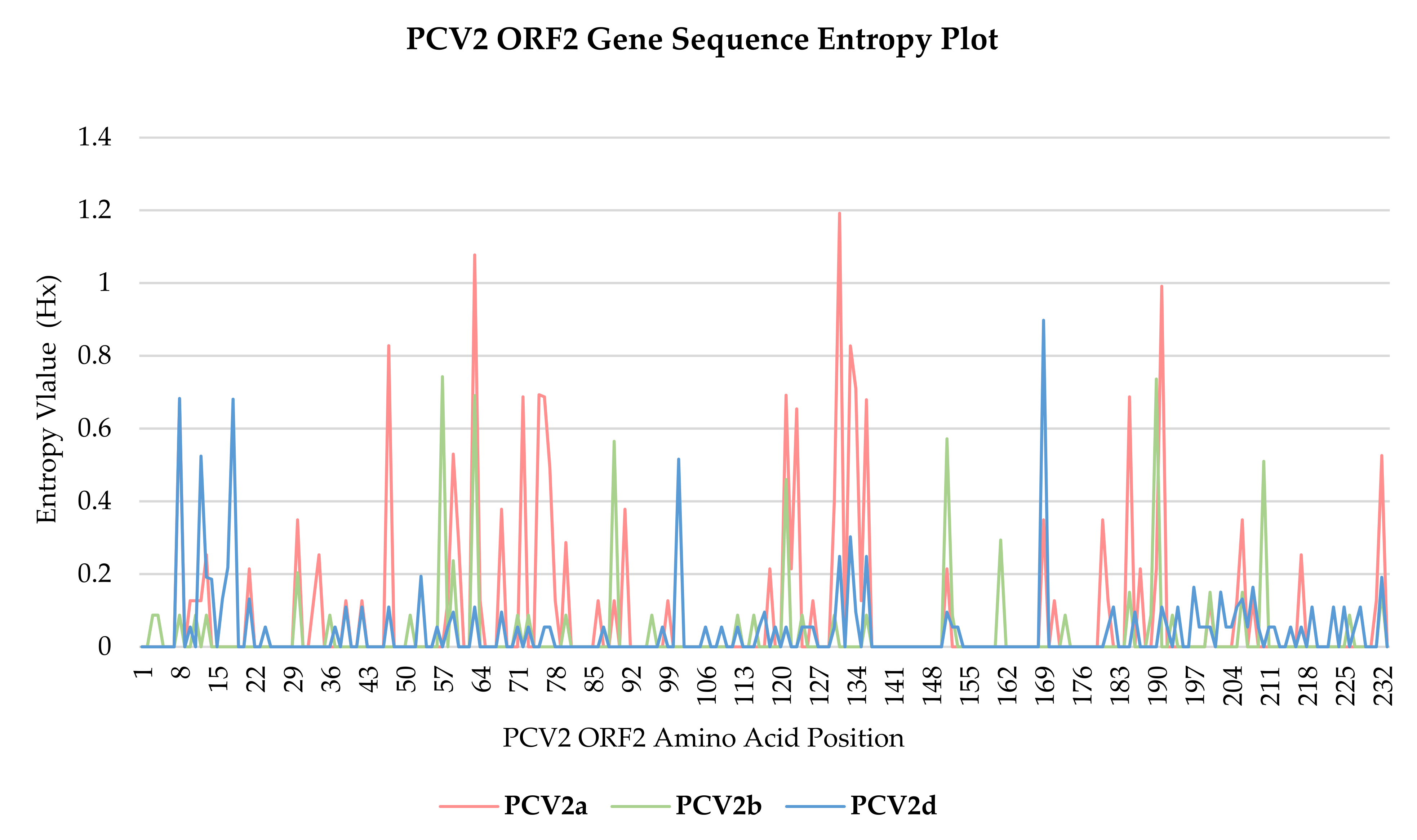
3.4. Nucleotide Sequence Polymorphism Analysis
3.5. Nucleotide Sequence Polymorphism Analysis
3.6. Rates of Substitution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tischer, I.; Gelderblom, H.; Vettermann, W.; Koch, M.A. A very small porcine virus with circular single-stranded DNA. Nature 1982, 295, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.G. Pathology of the post-weaning multisystemic wasting syndrome of pigs. In Proceedings of the Western Canadian Association of Swine Practitioners, Saskatoon, SK, Canada, 18–19 October 1996; Volume 1996, pp. 22–25. [Google Scholar]
- Harding, J.C. Post-weaning multisystemic wasting syndrome: Preliminary epidemiology and clinical findings. In Proceedings of the Western Canadian Association of Swine Practitioners, Saskatoon, SK, Canada, 18–19 October 1996; Volume 1996, p. 21. [Google Scholar]
- Ellis, J.; Hassard, L.; Clark, E.; Harding, J.; Allan, G.; Willson, P.; Strokappe, J.; Martin, K.; McNeilly, F.; Meehan, B.; et al. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can. Vet. J. 1998, 39, 44. [Google Scholar]
- Porcine Circovirus Associated Disease (PCVAD) Case Definition [Internet]. 2006. Available online: http://www.aasp.org/aasv/position-PCVAD.htm (accessed on 20 April 2022).
- Palinski, R.; Piñeyro, P.; Shang, P.; Yuan, F.; Guo, R.; Fang, Y.; Byers, E.; Hause, B.M. A novel porcine circovirus distantly related to known circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. J. Virol. 2017, 91, e01879-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Hu, W.Q.; Li, J.Y.; Liu, T.N.; Zhou, J.Y.; Opriessnig, T.; Xiao, C.T. Novel circovirus species identified in farmed pigs designated as Porcine circovirus 4, Hunan province, China. Transbound. Emerg. Dis. 2020, 67, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.; Li, L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meehan, B.M.; Creelan, J.L.; McNulty, M.S.; Todd, D. Sequence of porcine circovirus DNA: Affinities with plant circoviruses. J. Gen. Virol. 1997, 78, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, M.; Halbur, P.G.; Gill, M.; Toth, T.E.; Meng, X.J. Genetic characterization of type 2 porcine circovirus (PCV-2) from pigs with postweaning multisystemic wasting syndrome in different geographic regions of North America and development of a differential PCR-restriction fragment length polymorphism assay to detect and differentiate between infections with PCV-1 and PCV-2. J. Clin. Microbiol. 2000, 38, 2494–2503. [Google Scholar]
- Nawagitgul, P.; Morozov, I.; Bolin, S.R.; Harms, P.A.; Sorden, S.D.; Paul, P.S. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J. Gen. Virol. 2000, 81, 2281–2287. [Google Scholar] [CrossRef]
- Mankertz, A.; Çaliskan, R.; Hattermann, K.; Hillenbrand, B.; Kurzendoerfer, P.; Mueller, B.; Schmitt, C.; Steinfeldt, T.; Finsterbusch, T. Molecular biology of Porcine circovirus: Analyses of gene expression and viral replication. Vet. Microbiol. 2004, 98, 81–88. [Google Scholar] [CrossRef]
- Liu, J.; Chen, I.; Kwang, J. Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J. Virol. 2005, 79, 8262–8274. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, Y.; Chen, I.; Lau, J.; He, F.; Lau, A.; Wang, Z.; Karuppannan, A.K.; Kwang, J. The ORF3 protein of porcine circovirus type 2 interacts with porcine ubiquitin E3 ligase Pirh2 and facilitates p53 expression in viral infection. J. Virol. 2007, 81, 9560–9567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meehan, B.M.; McNeilly, F.; Todd, D.; Kennedy, S.; Jewhurst, V.A.; Ellis, J.A.; Hassard, L.E.; Clark, E.G.; Haines, D.M.; Allan, G.M. Characterization of novel circovirus DNAs associated with wasting syndromes in pigs. J. Gen. Virol. 1998, 79, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Morozov, I.; Sirinarumitr, T.; Sorden, S.D.; Halbur, P.G.; Morgan, M.K.; Yoon, K.J.; Paul, P.S. Detection of a novel strain of porcine circovirus in pigs with postweaning multisystemic wasting syndrome. J. Clin. Microbiol. 1998, 36, 2535–2541. [Google Scholar] [CrossRef] [Green Version]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Giannitti, F.; Rossow, S.; Marthaler, D.; Knutson, T.P.; Li, L.; Deng, X.; Resende, T.; Vannucci, F.; Delwart, E. Detection of a novel circovirus PCV3 in pigs with cardiac and multi-systemic inflammation. Virol. J. 2016, 13, 184. [Google Scholar] [CrossRef] [Green Version]
- Olvera, A.; Cortey, M.; Segales, J. Molecular evolution of porcine circovirus type 2 genomes: Phylogeny and clonality. Virology 2007, 357, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.; Wang, X.; Dvorak, C.M.; Marthaler, D.; Murtaugh, M.P. Diagnostic phylogenetics reveals a new Porcine circovirus 2 cluster. Virus Res. 2016, 217, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Afghah, Z.; Webb, B.; Meng, X.J.; Ramamoorthy, S. Ten years of PCV2 vaccines and vaccination: Is eradication a possibility? Vet. Microbiol. 2017, 206, 21–28. [Google Scholar] [CrossRef]
- Firth, C.; Charleston, M.A.; Duffy, S.; Shapiro, B.; Holmes, E.C. Insights into the evolutionary history of an emerging livestock pathogen: Porcine circovirus 2. J. Virol. 2009, 83, 12813–12821. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L.; Piontkivska, H. Nucleotide sequence polymorphism in circoviruses. Infect. Genet. Evol. 2008, 8, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Segalés, J. Porcine circovirus 2 (PCV-2) genotype update and proposal of a new genotyping methodology. PLoS ONE 2018, 13, e0208585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, S.; Toung, O.P.; Yee, P.L.; Yew, T.D.; Yoon, C.P.; Keong, L.B. Genetic characterization of Porcine Circovirus 2 found in Malaysia. Virol. J. 2011, 8, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.Y.; Opaskornkul, K.; Thanawongnuwech, R.; Arshad, S.S.; Hassan, L.; Ooi, P.T. First molecular detection and complete sequence analysis of porcine circovirus type 3 (PCV3) in Peninsular Malaysia. PLoS ONE 2020, 15, e0235832. [Google Scholar] [CrossRef]
- Fort, M.; Olvera, A.; Sibila, M.; Segalés, J.; Mateu, E. Detection of neutralizing antibodies in postweaning multisystemic wasting syndrome (PMWS)-affected and non-PMWS-affected pigs. Vet. Microbiol. 2007, 125, 44–55. [Google Scholar] [CrossRef] [Green Version]
- IBM. SPSS Statistics for Windows, Version 24.0; IBM: Armonk, NY, USA, 2016. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Franzo, G.; Cortey, M.; Olvera, A.; Novosel, D.; De Castro, A.M.; Biagini, P.; Segalés, J.; Drigo, M. Revisiting the taxonomical classification of Porcine Circovirus type 2 (PCV2): Still a real challenge. Virol. J. 2015, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Litwin, S.; Jores, R. Shannon information as a measure of amino acid diversity. In Theoretical and Experimental Insights into Immunology; Springer: Berlin/Heidelberg, Germany, 1992; pp. 279–287. [Google Scholar]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the Skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Grau-Roma, L.; Crisci, E.; Sibila, M.; Lopez-Soria, S.; Nofrarias, M.; Cortey, M.; Fraile, L.; Olvera, A.; Segalés, J. A proposal on porcine circovirus type 2 (PCV2) genotype definition and their relation with postweaning multisystemic wasting syndrome (PMWS) occurrence. Vet. Microbiol. 2008, 128, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Hassuzana, K.; Sharifah, S.H.; Siah, M.H.; Zuraidah, O.; Fauad, T.R. The use of restriction fragment length polymorphism technique to identify PMWS in pigs. In Proceedings of the 11th International Conference of the Association of Institutions for Tropical Veterinary Medicine, Petaling Jaya, Malaysia, 23–27 August 2004; p. 238. [Google Scholar]
- Ooi, P.T.; Choo, P.Y.; Hii, D.O.; Shahirudin, S.; Seetha, J.; Lim, B.K. Identification of porcine circovirus type 2 from pigs with postweaning multisystemic wasting syndrome in Malaysia. In Proceedings of the 19th Veterinary Malaysia Association Scientific Congress, Kuala Lumpur, Malaysia, 3–5 August 2007; pp. 109–111. [Google Scholar]
- Calsamiglia, M.; Segalés, J.; Quintana, J.; Rosell, C.; Domingo, M. Detection of porcine circovirus types 1 and 2 in serum and tissue samples of pigs with and without postweaning multisystemic wasting syndrome. J. Clin. Microbiol. 2002, 40, 1848–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madec, F.; Rose, N.; Grasland, B.; Cariolet, R.; Jestin, A. Post-weaning multisystemic wasting syndrome and other PCV2-related problems in pigs: A 12-year experience. Transbound. Emerg. Dis. 2008, 55, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Grau-Roma, L.; Fraile, L.; Segalés, J. Recent advances in the epidemiology, diagnosis and control of diseases caused by porcine circovirus type 2. Vet. J. 2011, 187, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J. Porcine circovirus type 2 (PCV2) infections: Clinical signs, pathology and laboratory diagnosis. Virus Res. 2012, 164, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Madson, D.M.; Ramamoorthy, S.; Kuster, C.; Pal, N.; Meng, X.J.; Halbur, P.G.; Opriessnig, T. Infectivity of porcine circovirus type 2 DNA in semen from experimentally-infected boars. Vet. Res. 2009, 40, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sibila, M.; Calsamiglia, M.; Segalés, J.; Blanchard, P.; Badiella, L.; Le Dimna, M.; Jestin, A.; Domingo, M. Use of a polymerase chain reaction assay and an ELISA to monitor porcine circovirus type 2 infection in pigs from farms with and without postweaning multisystemic wasting syndrome. Am. J. Vet. Res. 2004, 65, 88–92. [Google Scholar] [CrossRef]
- Grau-Roma, L.; Hjulsager, C.K.; Sibila, M.; Kristensen, C.S.; López-Soria, S.; Enøe, C.; Casal, J.; Bøtner, A.; Nofrarías, M.; Bille-Hansen, V.; et al. Infection, excretion and seroconversion dynamics of porcine circovirus type 2 (PCV2) in pigs from post-weaning multisystemic wasting syndrome (PMWS) affected farms in Spain and Denmark. Vet. Microbiol. 2009, 135, 272–282. [Google Scholar] [CrossRef]
- Sorden, S.D. Update on porcine circovirus and postweaning multisystemic wasting syndrome (PMWS). J. Swine Health Prod. 2000, 8, 133–136. [Google Scholar]
- Sanchez, R.; Nauwynck, H.; Pensaert, M. Serological survey of porcine circovirus 2 antibodies in domestic and feral pig populations in Belgium. In Proceedings of the ssDNA Viruses Plants, Birds, Pigs and Primates (ESVV) Meeting, St. Malo, France, 24–27 September 2001; p. 122. [Google Scholar]
- Vicente, J.; Segalés, J.; Höfle, U.; Balasch, M.; Plana-Durán, J.; Domingo, M.; Gortázar, C. Epidemiological study on porcine circovirus type 2 (PCV2) infection in the European wild boar (Sus scrofa). Vet. Res. 2004, 35, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Knell, S.; Willems, H.; Hertrampf, B.; Reiner, G. Comparative genetic characterization of porcine circovirus type 2 samples from German wild boar populations. Vet. Microbiol. 2005, 109, 169–177. [Google Scholar] [CrossRef]
- Cságola, A.; Kecskeméti, S.; Kardos, G.; Kiss, I.; Tuboly, T. Genetic characterization of type 2 porcine circoviruses detected in Hungarian wild boars. Arch. Virol. 2006, 151, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, K.; Bartova, E.; Machova, J. Antibodies to selected viral disease agents in wild boars from the Czech Republic. J. Wildl. Dis. 2008, 44, 777–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofia, M.; Billinis, C.; Psychas, V.; Birtsas, P.; Sofianidis, G.; Leontides, L.; Knowles, N.; Spyrou, V. Detection and genetic characterization of porcine circovirus 2 isolates from the first cases of postweaning multisystemic and wasting syndrome in wild boars in Greece. J. Wildl. Dis. 2008, 44, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrini, S.; Barocci, S.; Gavaudan, S.; Villa, R.; Briscolini, S.; Sabbatini, M.; Mattozzi, C.; Barchiesi, F.; Salamida, S.; Ferrari, M.; et al. Detection of porcine circovirus type 2 (PCV2) from wild boars in central Italy. Eur. J. Wildl. Res. 2009, 55, 465–469. [Google Scholar] [CrossRef]
- Turcitu, M.A.; Wellenberg, G.J.; Barboi, G.; Codreanu, M.D.; Vuta, V.B.; Nicolae, S.; Barbuceanu, F.; Coste, H.; Cioranu, R. Genetic diversity of porcine circovirus type 2 (PCV2) in the Romanian wild boar population. Res. Vet. Sci. 2011, 91, e103–e106. [Google Scholar] [CrossRef]
- Fabisiak, M.; Szczotka, A.; Podgórska, K.; Stadejek, T. Prevalence of infection and genetic diversity of porcine circovirus type 2 (PCV2) in wild boar (Sus scrofa) in Poland. J. Wildl. Dis. 2012, 48, 612–618. [Google Scholar] [CrossRef] [Green Version]
- An, D.J.; Lim, S.I.; Kim, Y.K.; Lee, H.K.; Cho, Y.Y.; Song, J.Y.; Hyun, B.H.; Park, B.K. Genetic characterization of porcine circovirus type 2 in the Korean wild boar population. Vet. Microbiol. 2014, 169, 147–153. [Google Scholar] [CrossRef]
- Ramos, N.; Mirazo, S.; Botto, G.; Teixeira, T.F.; Cibulski, S.P.; Castro, G.; Cabrera, K.; Roehe, P.M.; Arbiza, J. High frequency and extensive genetic heterogeneity of TTSuV1 and TTSuVk2a in PCV2-infected and non-infected domestic pigs and wild boars from Uruguay. Vet. Microbiol. 2018, 224, 78–87. [Google Scholar] [CrossRef]
- Nisavic, J.; Milic, N.; Radalj, A.; Mirilovic, M.; Vejnovic, B.; Cosic, M.; Knezevic, A.; Veljovic, L.; Zivulj, A. Detection and characterisation of porcine circoviruses in wild boars in northeastern Serbia. Veterinární Med. 2022, 67, 131–137. [Google Scholar] [CrossRef]
- Lipej, Z.; Segalés, J.; Jemeršić, L.; Olvera, A.; Roić, B.; Novosel, D.; Mihaljević, Ž.; Manojlović, L. First description of postweaning multisystemic wasting syndrome (PMWS) in wild boar (Sus scrofa) in Croatia and phylogenetic analysis of partial PCV2 sequences. Acta Vet. Hung. 2007, 55, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Fons, F.; Segalés, J.; Gortázar, C. A review of viral diseases of the European wild boar: Effects of population dynamics and reservoir rôle. Veter. J. 2008, 176, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Rudova, N.; Buttler, J.; Kovalenko, G.; Sushko, M.; Bolotin, V.; Muzykina, L.; Zinenko, O.; Stegniy, B.; Dunaiev, Y.; Sytiuk, M.; et al. Genetic Diversity of Porcine Circovirus 2 in Wild Boar and Domestic Pigs in Ukraine. Viruses 2022, 14, 924. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.R.; Bowen, R.A.; Bosco-Lauth, A.M. Zoonotic pathogens from feral swine that pose a significant threat to public health. Transbound. Emerg. Dis. 2018, 65, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Kim, K.S.; Ranyuk, M.; Babaev, E.; Voloshina, I.; Bayarlkhagva, D.; Chong, J.-R.; Ishiguro, N.; Yu, L.; Min, M.-S.; et al. Asia-wide phylogeography of wild boar (Sus scrofa) based on mitochondrial DNA and Y-chromosome: Revising the migration routes of wild boar in Asia. PLoS ONE 2020, 15, e0238049. [Google Scholar] [CrossRef]
- Pacini, M.; Forzan, M.; Cilia, G.; Bertelloni, F.; Fratini, F.; Mazzei, M. Detection and Characterization of Viral Pathogens Associated with Reproductive Failure in Wild Boars in Central Italy. Animals 2021, 11, 304. [Google Scholar] [CrossRef]
- Schulze, C.; Neumann, G.; Hlinak, A.; Segales, J.; Calsamiglia, M.; Domingo, M. Identification of postweaning multisystemic wasting syndrome in European wild boar (Sus scrofa). Veter. Rec. 2004, 154, 694–696. [Google Scholar] [CrossRef]
- Toplak, I.; Grom, J.; Hostnik, P.; Barlič-Maganja, D. Phylogenetic analysis of type 2 porcine circoviruses identified in wild boar in Slovenia. Veter. Rec. 2004, 155, 178–180. [Google Scholar] [CrossRef]
- Corner, L. The role of wild animal populations in the epidemiology of tuberculosis in domestic animals: How to assess the risk. Veter. Microbiol. 2006, 112, 303–312. [Google Scholar] [CrossRef]
- Wobeser, G.A. Disease and epizootiology-Basic principles. In Investigation and Management of Disease in Wild Animals, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 393–451. [Google Scholar]
- Franzo, G.; Tinello, S.; Grassi, L.; Tucciarone, C.M.; Legnardi, M.; Cecchinato, M.; Dotto, G.; Mondin, A.; Martini, M.; Pasotto, D.; et al. Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries. Pathogens 2020, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Albina, E.; Mesplede, A.; Chenut, G.; Le Potier, M.F.; Bourbao, G.; Le Gal, S.; Leforban, Y. A serological survey on classical swine fever (CSF), Aujeszky’s disease (AD) and porcine reproductive and respiratory syndrome (PRRS) virus infections in French wild boars from 1991 to 1998. Vet. Microbiol. 2000, 77, 43–57. [Google Scholar] [CrossRef]
- Laddomada, A. Incidence and control of CSF in wild boar in Europe. Vet. Microbiol. 2000, 73, 121–130. [Google Scholar] [CrossRef]
- Vicente, J.; León-Vizcaíno, L.; Gortázar, C.; Cubero, M.J.; González, M.; Martín-Atance, P. Antibodies to selected viral and bacterial pathogens in European wild boars from southcentral Spain. J. Wildl. Dis. 2002, 38, 649–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupancić, Z.; Jukić, B.; Lojkić, M.; Cac, Z.; Jemeršić, L.; Starešina, V. Prevalence of antibodies to classical swine fever, Aujeszky’s disease, porcine reproductive and respiratory syndrome, and bovine viral diarrhoea viruses in wild boars in Croatia. J. Vet. Med. Ser. B 2002, 49, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Sauter-Louis, C.; Conraths, F.J.; Probst, C.; Blohm, U.; Schulz, K.; Sehl, J.; Fischer, M.; Forth, J.H.; Zani, L.; Depner, K.; et al. African swine fever in wild boar in Europe—A review. Viruses 2021, 13, 1717. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Zhang, X.; Liu, X.; Ren, L. Co-infection of swine with porcine circovirus type 2 and other swine viruses. Viruses 2019, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Ruan, H.; Qiao, S.; Deng, R.; Zhang, G. Co-infection status of porcine circoviruses (PCV2 and PCV3) and porcine epidemic diarrhea virus (PEDV) in pigs with watery diarrhea in Henan province, central China. Microb. Pathog. 2020, 142, 104047. [Google Scholar] [CrossRef]
- Visuthsak, W.; Woonwong, Y.; Thanantong, N.; Poolperm, P.; Boonsoongnern, A.; Ratanavanichrojn, N.; Jirawattanapong, P.; Soda, N.; Kaminsonsakul, T.; Phuttapatimok, S.; et al. PCV3 in Thailand: Molecular epidemiology and relationship with PCV2. Transbound. Emerg. Dis. 2021, 68, 2980–2989. [Google Scholar] [CrossRef]
- Xu, T.; Hou, C.Y.; Zhang, Y.H.; Li, H.X.; Chen, X.M.; Pan, J.J.; Chen, H.Y. Simultaneous detection and genetic characterization of porcine circovirus 2 and 4 in Henan province of China. Gene 2022, 808, 145991. [Google Scholar] [CrossRef]
- Kim, H.R.; Park, Y.R.; Lim, D.R.; Park, M.J.; Park, J.Y.; Kim, S.H.; Lee, K.K.; Lyoo, Y.S.; Park, C.K. Multiplex real-time polymerase chain reaction for the differential detection of porcine circovirus 2 and 3. J. Virol. Methods 2017, 250, 11–16. [Google Scholar] [CrossRef]
- Li, X.; Qiao, M.; Sun, M.; Tian, K. A Duplex Real-Time PCR Assay for the Simultaneous Detection of Porcine Circovirus 2 and Circovirus. Virol. Sin. 2018, 33, 181–186. [Google Scholar]
- Jiang, H.; Wang, D.; Wang, J.; Zhu, S.; She, R.; Ren, X.; Tian, J.; Quan, R.; Hou, L.; Li, Z.; et al. Induction of Porcine Dermatitis and Nephropathy Syndrome in Piglets by Infection with Porcine Circovirus Type. J. Virol. 2019, 93, e02045-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.J.; de Arce, H.D.; Cortey, M.; Domínguez, P.; Percedo, M.I.; Perera, C.L.; Tarradas, J.; Frías, M.T.; Segalés, J.; Ganges, L.; et al. Phylogenetic networks to study the origin and evolution of porcine circovirus type 2 (PCV2) in Cuba. Veter. Microbiol. 2011, 151, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Cortey, M.; Segalés, J.; Hughes, J.; Drigo, M. Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol. Phylogenetics Evol. 2016, 100, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Hesse, R.; Kerrigan, M.; Rowland, R.R. Evidence for recombination between PCV2a and PCV2b in the field. Virus Res. 2008, 132, 201–207. [Google Scholar] [CrossRef]
- Cai, L.; Han, X.; Ni, J.; Yu, X.; Zhou, Z.; Zhai, X.; Chen, X.; Tian, K. Natural recombinants derived from different patterns of recom-bination between two PCV2b parental strains. Virus Res. 2011, 158, 281–288. [Google Scholar] [CrossRef]
- Zhai, S.-L.; Chen, S.-N.; Wei, Z.-Z.; Zhang, J.-W.; Huang, L.; Lin, T.; Yue, C.; Ran, D.-L.; Yuan, S.-S.; Wei, W.-K.; et al. Co-existence of multiple strains of porcine circovirus type 2 in the same pig from China. Virol. J. 2011, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Chiou, M.-T.; Lin, C.-N.; Yang, C.-Y.; Su, G.-S.; Lin, C.-F.; Chang, T.-C. Genotypic Change and Phylogenetic Analysis of Porcine Circovirus Type 2 in Taiwanese Pig Herds. J. Veter. Med. Sci. 2012, 74, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K.; Lager, K.M.; Kohutyuk, O.I.; Vincent, A.L.; Henry, S.C.; Baker, R.B.; Rowland, R.R.; Dunham, A.G. Detection of two porcine circovirus type 2 genotypic groups in United States swine herds. Arch. Virol. 2007, 152, 1035–10446. [Google Scholar] [CrossRef]
- Carman, S.; Cai, H.Y.; DeLay, J.; Youssef, S.A.; McEwen, B.J.; Gagnon, C.A.; Tremblay, D.; Hazlett, M.; Lusis, P.; Fairles, J.; et al. The emergence of a new strain of porcine circovirus-2 in Ontario and Quebec swine and its association with severe porcine circovirus associated disease—2004–2006. Can. J. Vet. Res. 2008, 72, 259–268. [Google Scholar]
- Dupont, K.; Nielsen, E.; Bækbo, P.; Larsen, L. Genomic analysis of PCV2 isolates from Danish archives and a current PMWS case–control study supports a shift in genotypes with time. Veter. Microbiol. 2008, 128, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Guo, X.; Ge, X.; Wang, Z.; Chen, Y.; Cha, Z.; Yang, H. Genetic variation analysis of Chinese strains of porcine circovirus type. Virus Res. 2009, 145, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Cortey, M.; Pileri, E.; Sibila, M.; Pujols, J.; Balasch, M.; Plana, J.; Segalés, J. Genotypic shift of porcine circovirus type 2 from PCV-2a to PCV-2b in Spain from 1985 to 2008. Veter. J. 2011, 187, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, C.A.; Tremblay, D.; Tijssen, P.; Venne, M.-H.; Houde, A.; Elahi, S.M. The emergence of porcine circovirus 2b genotype (PCV-2b) in swine in Canada. Can. Vet. J. 2007, 48, 811. [Google Scholar]
- Xiao, C.; Halbur, P.G.; Opriessnig, T. Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J. Gen. Virol. 2015, 96, 1830–1841. [Google Scholar] [CrossRef]
- Department of Veterinary Services Malaysia (DVS Malaysia). Livestock Statistics [Internet]. Available online: http://www.dvs.gov.my/index.php/pages/view/2758?mid=42 (accessed on 30 December 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCV2 PCR Detection Rate | Domestic Pig | Abattoir | Wild Boar | |||
|---|---|---|---|---|---|---|
| Northern Malaysia | Central Malaysia | Southern Malaysia | Overall | |||
| % PCR Positive (Animals) | 93.10% (27/29) | 82.76% (24/29) | 71.43% (15/21) | 83.54% (66/79) | 94.74% (18/19) | 100.00% (28/28) |
| % PCR Positive (Farms) | 92.86% (13/14) | 85.71% (12/14) | 66.67% (6/9) | 83.78% (31/37) | - | - |
| Production Age Group | % PCR Positive (Number of Positive Animals/Tested Animal) |
|---|---|
| Fetus | 100% (7/7) |
| Piglet | 100% (2/2) |
| Weaner | 80.36% (45/56) |
| Grower | 83.33% (10/12) |
| Sow | 100% (2/2) |
| Detection Status of Tested Pig (Number of Individuals) | PCV2 Positive | PCV2 Negative |
|---|---|---|
| PCV3 Positive | 21 | 1 |
| PCV3 Negative | 39 | 12 |
| Marker Nucleotide Position | 157 A | 162 T | 513 C | 585 T | 643 A | |
|---|---|---|---|---|---|---|
| Malaysian PCV2d Sequences | ||||||
| KL44/OM524605 | A | T | A | T | A | |
| SL37/OM524602 | A | T | A | T | A | |
| PG201L/OM524626 | A | T | A | T | A | |
| PG242L/OM524638 | A | T | A | T | A | |
| PG39L/OM524603 | A | T | A | T | A | |
| PW199L/OM524625 | A | T | A | T | A | |
| WB2/OM524650 | T | A | C | T | A | |
| WB5/OM524653 | A | T | A | T | A | |
| WB10/OM524657 | T | T | C | T | A | |
| % Sequences not matching marker nucleotides | 2.63% (2/76) | 1.32% (1/76) | 9.21% (7/76) | 0.00% | 0.00% | |
| PCV2 Genotype | Tests of Neutrality | |||||
|---|---|---|---|---|---|---|
| Tajima’s D | Fu and Li’s D | Fu and Li’s F | ||||
| Test Statistic | Statistical Significance, p-Value | Test Statistic | Statistical Significance, p-Value | Test Statistic | Statistical Significance, p-Value | |
| PCV2a | −1.2028 | >0.10 | −2.2422 | 0.10 > p > 0.05 | −2.2331 | 0.10 > p > 0.05 |
| PCV2b | −2.1293 | <0.05 | −3.3600 | <0.05 | −3.4549 | <0.02 |
| PCV2d | −2.5171 | <0.001 | −5.8800 | <0.02 | −5.2945 | <0.02 |
| PCV2 Genotype | Positive Selection | Negative Selection | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Positively Selected Codon | Selection Pressure Inference Methods | Statistical Consensus across Methods | Total Negatively Selected Codons | Selection Pressure Inference Methods | Statistical Consensus across Methods | |||||||||||
| FUBAR | FEL | SLAC | MEME | n = 1 | n = 2 | n = 3 | n = 4 | FUBAR | FEL | SLAC | n = 1 | n = 2 | n = 3 | |||
| PCV2a | 3.86% (9/233) | 3 | 1 | 0 | 7 | 8 | 0 | 1 | 0 | 13.73% (32/233) | 29 | 31 | 14 | 4 | 14 | 14 |
| PCV2b | 1.72% (4/233) | 3 | 0 | 0 | 1 | 4 | 0 | 0 | 0 | 13.73% (32/233) | 30 | 22 | 9 | 12 | 11 | 9 |
| PCV2d | 3.00% (7/233) | 3 | 1 | 1 | 6 | 5 | 1 | 0 | 1 | 13.30% (31/233) | 27 | 26 | 9 | 9 | 13 | 9 |
| PCV2 Genotype | ORF2 aa Site with Hx Values > 0.0 | Lowest Hx Value | Highest Hx Value | Standard Deviation, σ |
|---|---|---|---|---|
| PCV2a (36) | 23.61% (55/233) | 0.127 | 1.192 | 0.208 |
| PCV2b (58) | 15.02% (35/233) | 0.087 | 0.743 | 0.111 |
| PCV2d (103) | 31.76% (74/233) | 0.055 | 0.898 | 0.108 |
| PCV2 Genotype | Mean Rate of Substitution (Substitution per Site per Year) | 95% Highest Posterior Density (HPD) | Effective Sample Size (ESS) |
|---|---|---|---|
| PCV2a | 4.746 × 10−4 | 8.463 × 10−4 | 246,638 |
| PCV2b | 6.571 × 10−4 | 1.024 × 10−3 | 255,628 |
| PCV2d | 2.111 × 10−3 | 2.925 × 10−3 | 68,174 |
| Combined analysis for all genotypes | 1.102 × 10−3 | 1.374 × 10−3 | 82,401 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.Y.; Thanawongnuwech, R.; Arshad, S.S.; Hassan, L.; Fong, M.W.C.; Ooi, P.T. Genotype Shift of Malaysian Porcine Circovirus 2 (PCV2) from PCV2b to PCV2d within a Decade. Animals 2022, 12, 1849. https://doi.org/10.3390/ani12141849
Tan CY, Thanawongnuwech R, Arshad SS, Hassan L, Fong MWC, Ooi PT. Genotype Shift of Malaysian Porcine Circovirus 2 (PCV2) from PCV2b to PCV2d within a Decade. Animals. 2022; 12(14):1849. https://doi.org/10.3390/ani12141849
Chicago/Turabian StyleTan, Chew Yee, Roongroje Thanawongnuwech, Siti Suri Arshad, Latiffah Hassan, Michelle Wai Cheng Fong, and Peck Toung Ooi. 2022. "Genotype Shift of Malaysian Porcine Circovirus 2 (PCV2) from PCV2b to PCV2d within a Decade" Animals 12, no. 14: 1849. https://doi.org/10.3390/ani12141849