
Comparative Analysis of Fecal Microbiota of Grazing Mongolian Cattle from Different Regions in Inner Mongolia, China

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
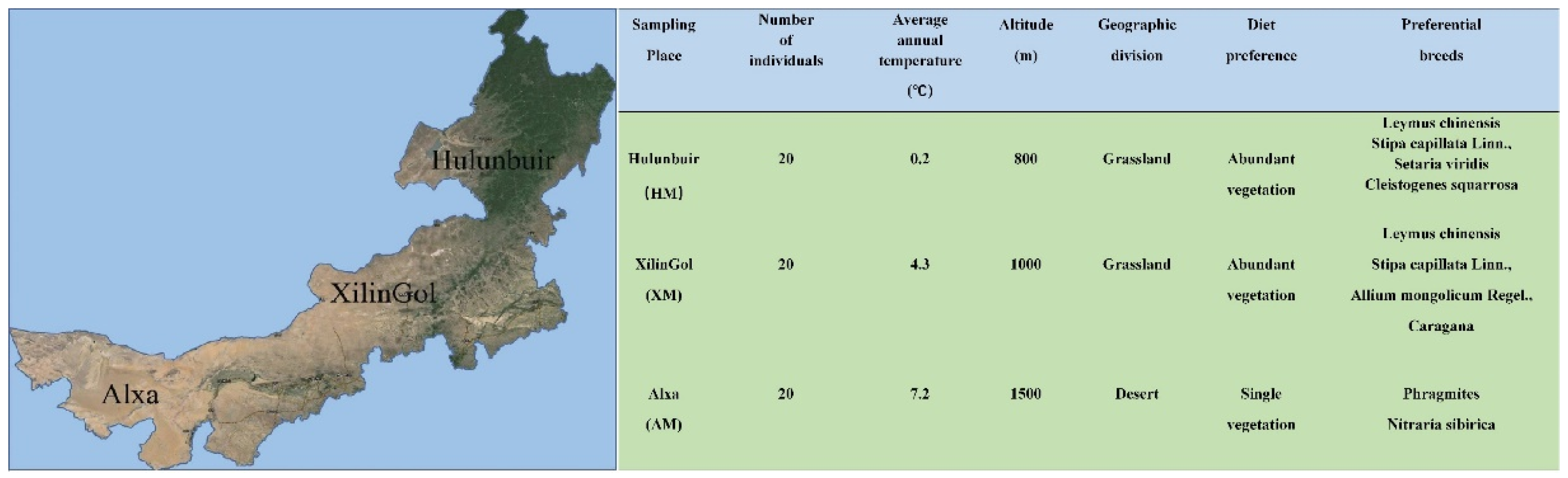
2.1. Sample Collection
2.2. Genome DNA Extraction
2.3. PCR Amplification and Hiseq Sequencing
2.4. Sequence Analysis
2.5. Data Statistics
3. Results
3.1. 16S rRNA Gene Sequencing
3.2. Microbial Diversity
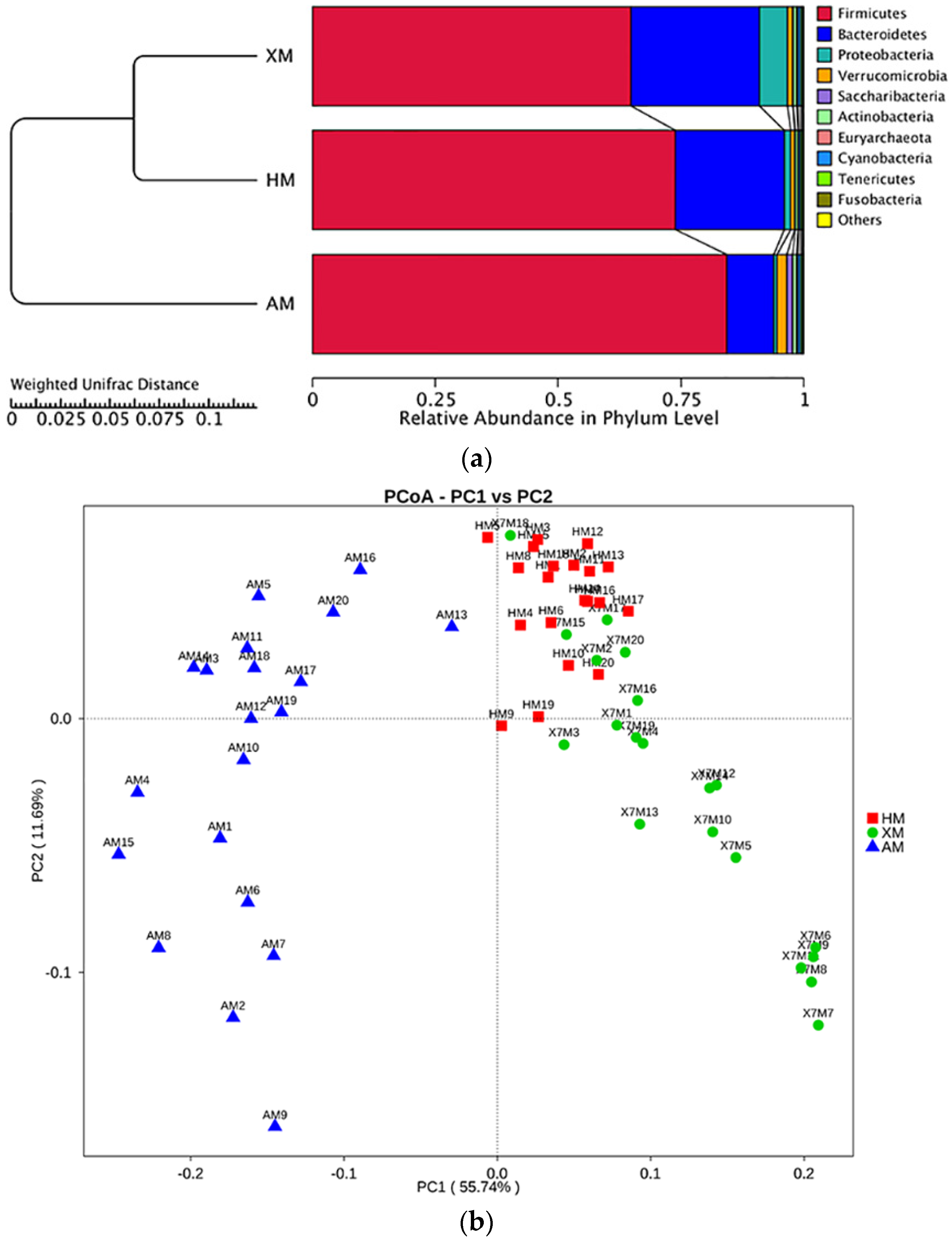
3.3. Beta Diversity
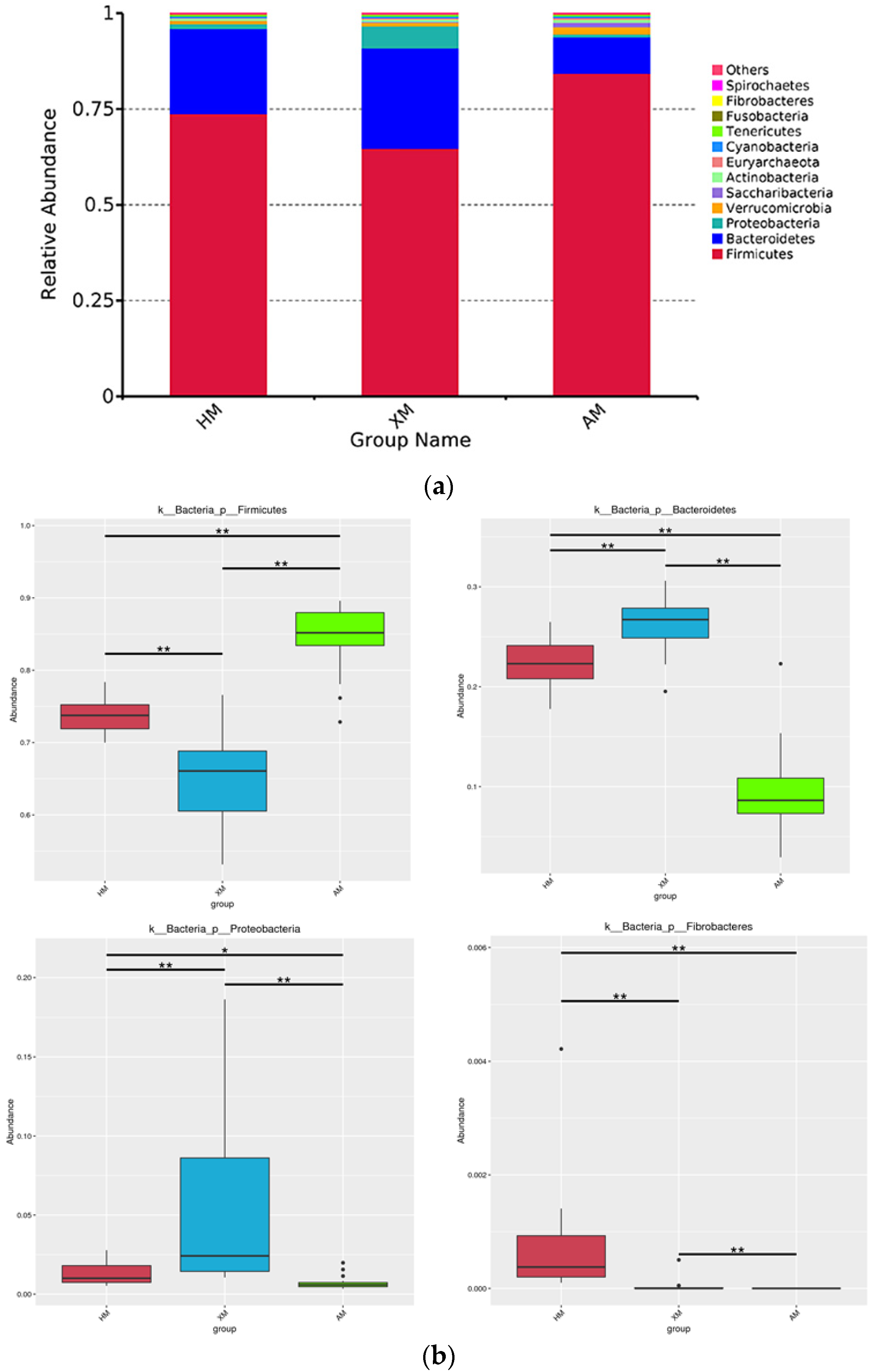
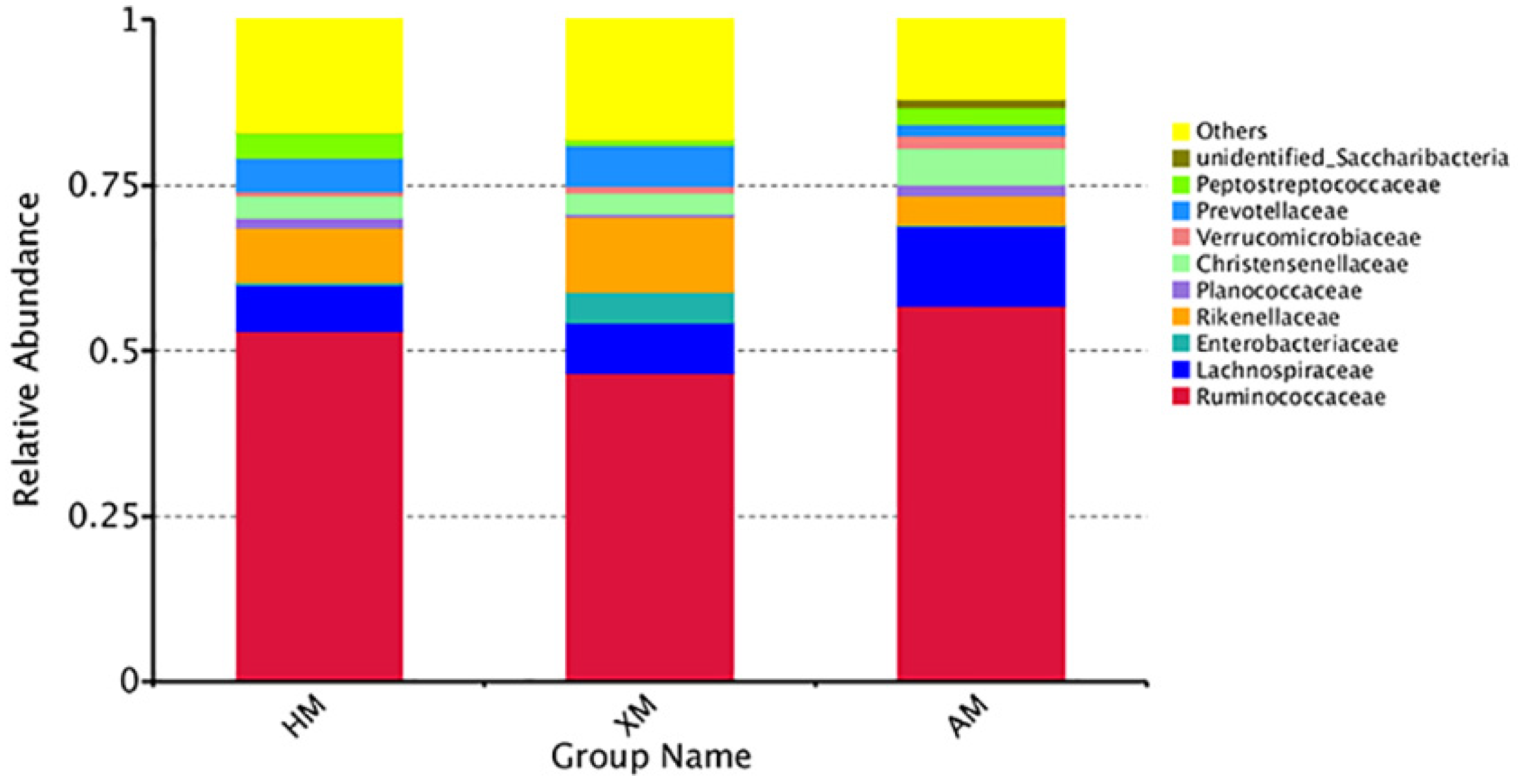
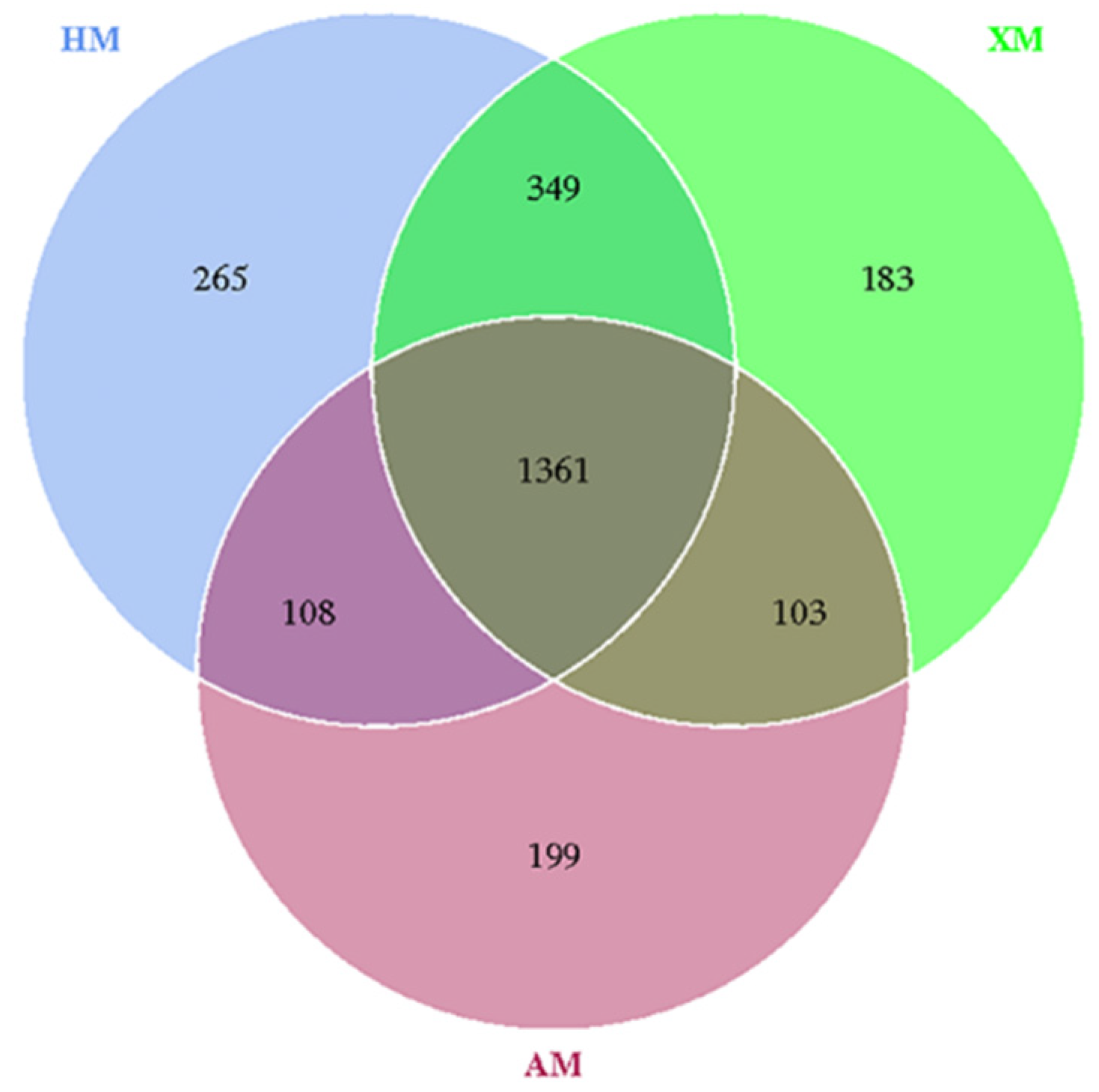
3.4. Bacterial Community Taxonomic Composition
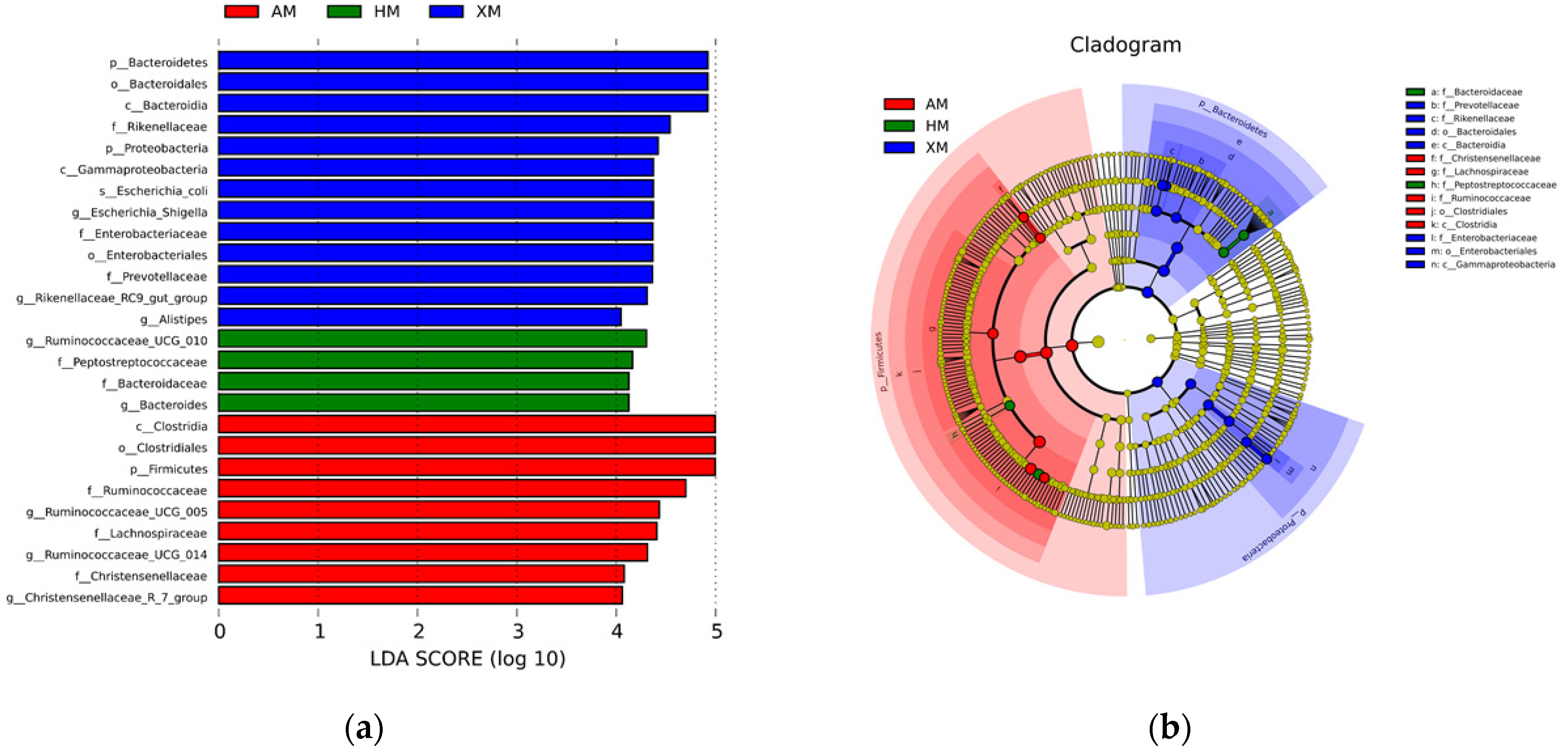
3.5. Microbial Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1079–U49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, R.A.; Young, V.B. Role of the intestinal microbiota in resistance to colonization by clostridium difficile. Gastroenterology 2014, 146, 1547–1553. [Google Scholar] [CrossRef] [Green Version]
- Org, E.; Parks, B.W.; Joo, J.W.J.; Emert, B.; Schwartzman, W.; Kang, E.Y.; Mehrabian, M.; Pan, C.; Knight, R.; Gunsalus, R.; et al. Genetic and environmental control of host-gut microbiota interactions. Genome. Res. 2015, 25, 1558–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, S.; Premathilake, H.; Desilva, U.; Shili, C.; Carter, S.; Pezeshki, A. Low protein-high carbohydrate diets alter energy balance, gut microbiota composition and blood metabolomics profile in young pigs. Sci. Rep. 2020, 10, 3318–3332. [Google Scholar] [CrossRef]
- Lan, D.L.; Ji, W.H.; Lin, B.S.; Chen, Y.B.; Huang, C.; Xiong, X.R.; Fu, M.; Mipam, T.D.; Ai, Y.; Zeng, B.; et al. Correlations between gut microbiota community structures of Tibetans and geography. Sci. Rep. 2017, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, B.C.; Tan, J.; Zhu, L.; Lou, D.; Cen, X. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol. Genet. Genom. 2017, 292, 407–414. [Google Scholar] [CrossRef]
- Yang, B.; Yan, S.; Chen, Y.; Ross, R.P.; Stanton, C.; Zhao, J.X.; Zhang, H.; Chen, W. Diversity of gut microbiota and bifidobacterial community of Chinese subjects of different ages and from different regions. Microorganisms 2020, 8, 1108. [Google Scholar] [CrossRef]
- Liang, M.; Li, Y.; Siriguleng; Surong, H.; He, J.; Le, H.; Wang, Z.; Guo, F.; Qiao, X.; Jirimutu. Comparative analysis of fecal microbial communities in cattle and Bactrian camels. PLoS ONE 2017, 12, e0173062. [Google Scholar] [CrossRef] [Green Version]
- Angelakis, E.; Bachar, D.; Yasir, M.; Musso, D.; Djossou, F.; Melenotte, C.; Robert, C.; Davoust, B.; Gaborit, B.; Azhar, E.I.; et al. Comparison of the gut microbiota of obese individuals from different geographic origins. N. Microbe. N. Infect 2019, 27, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wang, R.; Luo, J.; Ren, F.; Gu, Z.L.; Zhao, Y.Q.; Zhao, L. The Core and distinction of the gut microbiota in Chinese populations across geography and ethnicity. Microorganisms 2020, 8, 1579. [Google Scholar] [CrossRef]
- Kabwe, M.H.; Vikram, S.; Mulaudzi, K.; Jansson, J.K.; Makhalanyame, T.P. The gut microbiota of rural and urban individuals is shaped by geography and lifestyle. BMC Microbiol. 2020, 20, 257–268. [Google Scholar] [CrossRef]
- Tang, J.N.; Zeng, Z.G.; Wang, H.N.; Yang, T.; Zhang, P.J.; Li, Y.L.; Zhang, A.Y.; Fan, W.Q.; Zhang, Y.; Yang, X.; et al. An effective method for isolation of DNA from pig feces and comparison of five different methods. J. Microbiol. Methods 2008, 75, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Uparse: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Native Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Han, H.; Li, Y.; Liu, Z.J.; Zhao, Y.R.; Fang, R.J.; Huang, X.G.; Zheng, J.; Ren, W.K.; Wu, F.; et al. Lysine restriction affects feed intake and amino acid metabolism via gut microbiome in piglets. Cell. Physiol. Biochem. 2017, 44, 1749–1761. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome. Biol 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippo, C.D.; Cavalieri, D.; Paola, M.D.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 4691–4696. [Google Scholar] [CrossRef] [Green Version]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun 2014, 5, 3654. [Google Scholar] [CrossRef]
- Zhang, J.C.; Guo, Z.; Xue, Z.S.; Sun, Z.H.; Zhang, M.H.; Wang, L.F.; Wang, G.Y.; Wang, F.; Xu, J.; Cao, H.F.; et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME. J 2015, 9, 1979–1990. [Google Scholar] [CrossRef]
- Lau, S.K.; Teng, J.L.; Chiu, T.H.; Chan, E.; Tsang, A.K.; Panagiotou, G.; Zhai, S.L.; Woo, P.C. Differential microbial communities of omnivorous and herbivorous cattle in southern China. Comput. Struct. Biotec. 2018, 16, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; Mckeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Brulc, J.M.; Antonopoulos, D.A.; Berg Miller, M.E.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.; Bono, J.L.; Desantis, T.Z.; Harhay, D.M.; Andersen, G.L.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl. Environ. Microb. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Shinkai, T.; Koike, S. Ecological and physiological characterization shows that Fibrobacter succinogenes is important in rumen fiber digestion—Review. Folia. Microbiol. 2008, 53, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, G.; Lagier, J.C.; Armougom, F.; Robert, C.; Audoly, G.; Papazian, L.; Raoult, D. High-level colonisation of the human gut by Verrucomicrobia following broad-spectrum antibiotic treatment. Int. J. Antmicrob. Agents 2013, 41, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Orpin, C.G.; Mathiesen, S.D.; Greenwood, Y.; Blix, A.S. Seasonal changes in the ruminal microflora of the high-arctic svalbard reindeer (rangifer tarandus platyrhynchus). Appl. Environ. Microbiol. 1985, 50, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Varel, V.H.; Dehority, B.A. Ruminal cellulolytic bacteria and protozoa from bison, cattle-bison hybrids, and cattle fed three alfalfa-corn diets. Appl. Environ. Microbiol. 1989, 55, 148–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharm. Therap. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Rogers, M.A.M.; Ruffin, M.T.; Schloss, P.D. The human gut microbiome as a screening tool for colorectal cancer. Cancer. Prev. Res 2014, 7, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.C.; Gurram, B.; Baldridge, M.T.; Richard, H.; Lam, V.; Luo, C.W.; Cao, Y.M.; Simpson, P.; Hayward, M.A.; Holtz, M.L.; et al. O-011 Paneth Cell Phenotypes define a subtype of pediatric Crohn’s disease through alterations in host-microbial interactions. Inflamm. Bowel. Dis. 2016, 22 (Suppl. 1). [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Observed_Species | Community Diversity | Community Richness | |||
|---|---|---|---|---|---|---|
| Shannon | Simpson | Chao1 | Ace | Goods_Coverage (%) | ||
| HM | 1144.300 ± 50.761 a | 8.001 ± 0.113 a | 0.985 ± 0.002 a | 1368.287 ± 92.585 a | 1359.763 ± 78.557 a | 98.70 ± 0.001 b |
| XM | 1096.950 ± 50.217 b | 7.884 ± 0.296 a | 0.982 ± 0.008 a | 1301.873 ± 71.984 b | 1302.415 ± 70.688 b | 98.70 ± 0.001 b |
| AM | 823.900 ± 70.532 c | 7.326 ± 0.324 b | 0.980 ± 0.006 b | 951.889 ± 197.518 c | 938.770 ± 100.786 c | 99.20 ± 0.001 a |
| p-Value | <0.0001 | <0.0001 | 0.03 | <0.0001 | <0.0001 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aricha, H.; Simujide, H.; Wang, C.; Zhang, J.; Lv, W.; Jimisi, X.; Liu, B.; Chen, H.; Zhang, C.; He, L.; et al. Comparative Analysis of Fecal Microbiota of Grazing Mongolian Cattle from Different Regions in Inner Mongolia, China. Animals 2021, 11, 1938. https://doi.org/10.3390/ani11071938
Aricha H, Simujide H, Wang C, Zhang J, Lv W, Jimisi X, Liu B, Chen H, Zhang C, He L, et al. Comparative Analysis of Fecal Microbiota of Grazing Mongolian Cattle from Different Regions in Inner Mongolia, China. Animals. 2021; 11(7):1938. https://doi.org/10.3390/ani11071938
Chicago/Turabian StyleAricha, Han, Huasai Simujide, Chunjie Wang, Jian Zhang, Wenting Lv, Xirnud Jimisi, Bo Liu, Hao Chen, Chen Zhang, Lina He, and et al. 2021. "Comparative Analysis of Fecal Microbiota of Grazing Mongolian Cattle from Different Regions in Inner Mongolia, China" Animals 11, no. 7: 1938. https://doi.org/10.3390/ani11071938