Estimation of Linkage Disequilibrium and Effective Population Size in Three Italian Autochthonous Beef Breeds


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
2.2. Genotyping and Quality Control
2.3. Genomic Relationship Matrix
2.4. Linkage Disequilibrium
2.5. Estimation of Historical and Contemporary Effective Population Size
3. Results
3.1. Quality Control and Genomic Relationship Matrix
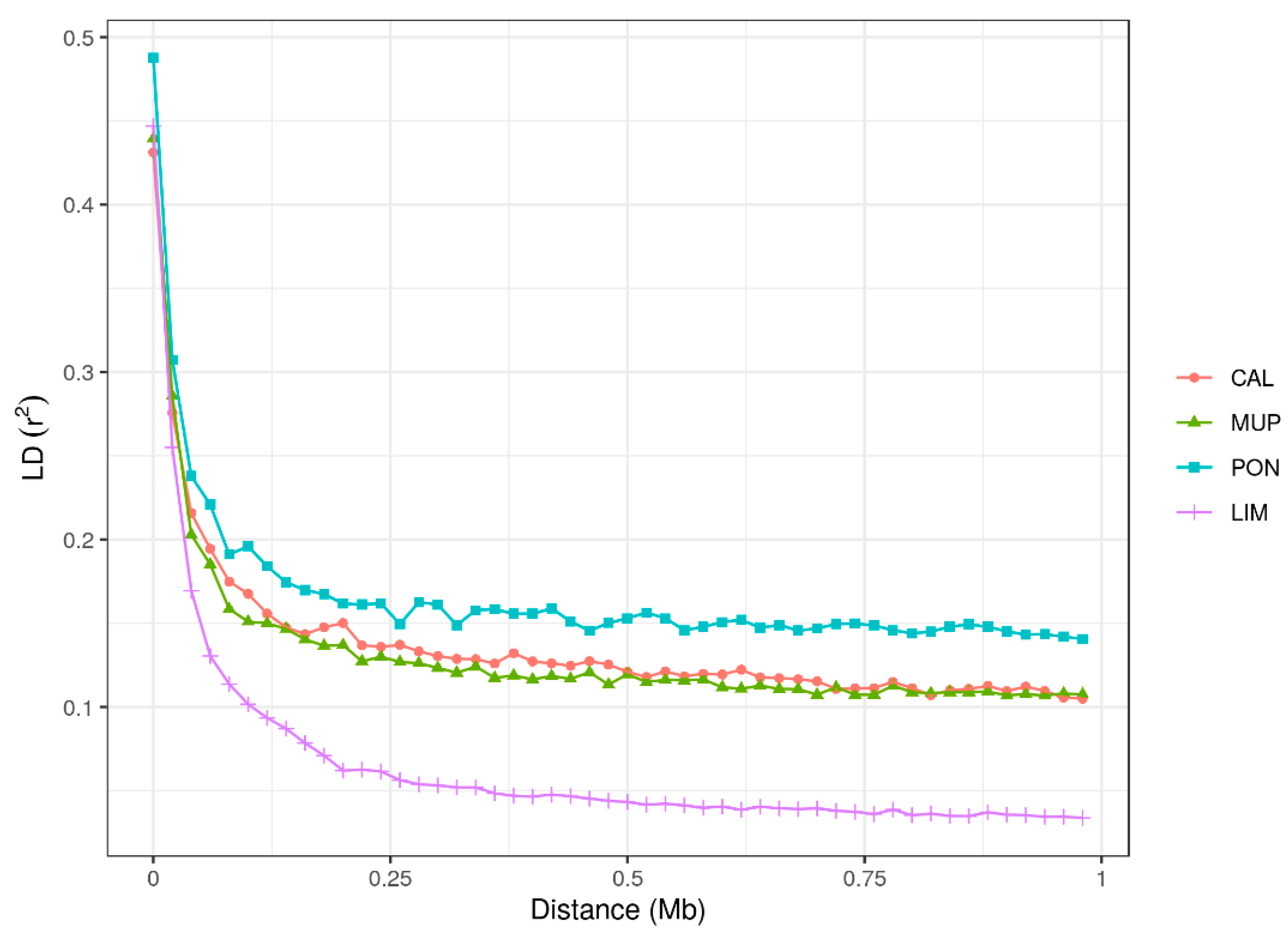
3.2. Linkage Disequilibrium
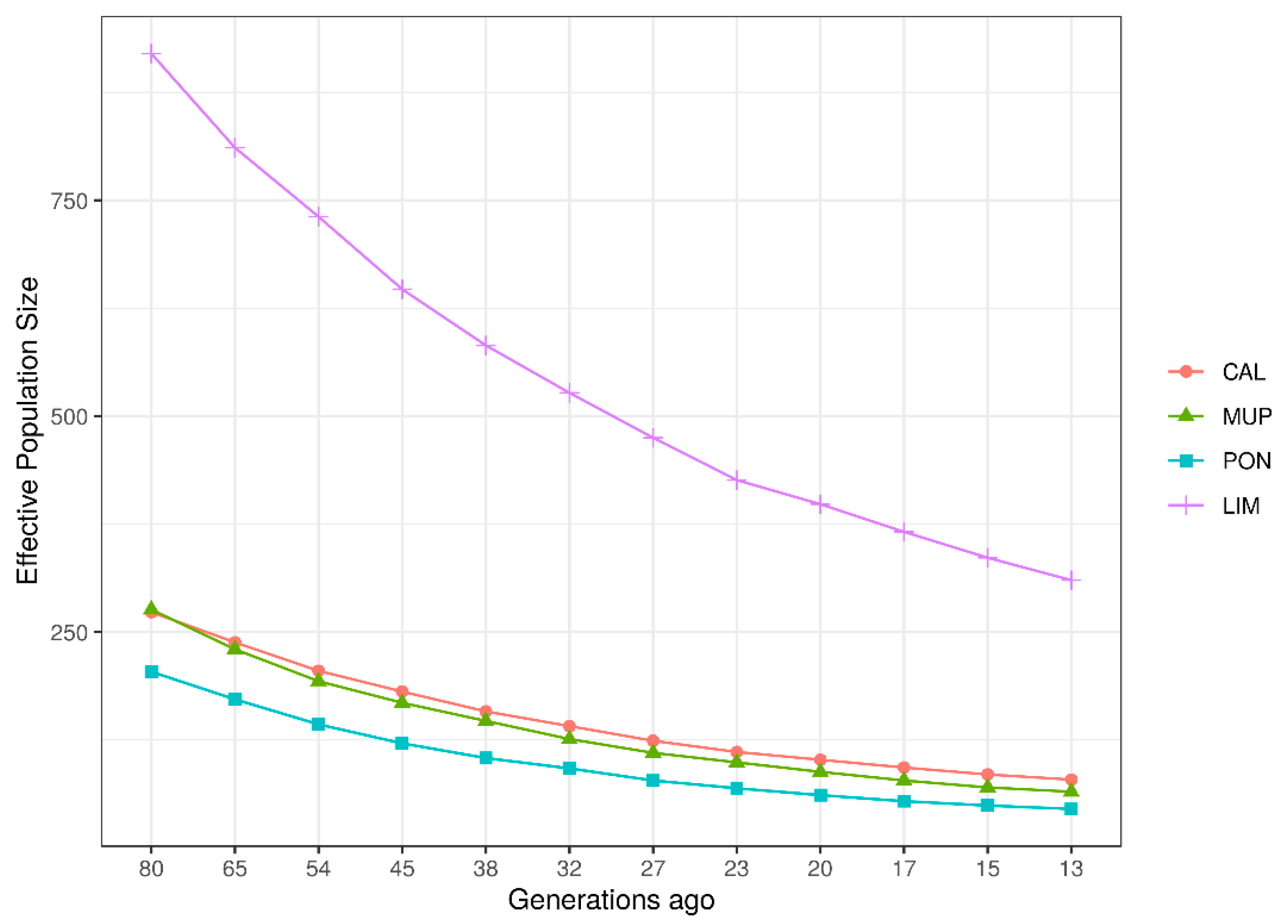
3.3. Estimation of Historical and Contemporary Effective Population Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bozzi, R.; Alvarez, I.; Crovetti, A.; Fernández, I.; De Petris, D.; Goyache, F. Assessing priorities for conservation in Tuscan cattle breeds using microsatellites. Anim. Int. J. Anim. Biosci. 2012, 6, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Negrini, R.; Milanesi, E.; Bozzi, R.; Pellecchia, M.; Ajmone-Marsan, P. Tuscany autochthonous cattle breeds: An original genetic resource investigated by AFLP markers. J. Anim. Breed. Genet. 2006, 123, 10–16. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Ciani, E.; Ajmone Marsan, P.; Bagnato, A.; Battaglini, L.; Bozzi, R.; Carta, A.; Catillo, G.; Cassandro, M.; Casu, S.; et al. Conservation status and historical relatedness of Italian cattle breeds. Genet. Sel. Evol. 2018, 50, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, G.; Mary-Huard, T.; Verrier, E.; Danvy, S.; Charvolin, E.; Danchin-Burge, C. Methods to estimate effective population size using pedigree data: Examples in dog, sheep, cattle and horse. Genet. Sel. Evol. 2013, 45, 1. [Google Scholar] [CrossRef] [Green Version]
- Palstra, F.P.; Ruzzante, D.E. Genetic estimates of contemporary effective population size: What can they tell us about the importance of genetic stochasticity for wild population persistence? Mol. Ecol. 2008, 17, 3428–3447. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.F.; Kimura, M. An introduction to population genetics theory. Introd. Popul. Genet. Theory 1970, 2, 174–208. [Google Scholar]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef]
- Fabbri, M.C.; Gonçalves de Rezende, M.P.; Dadousis, C.; Biffani, S.; Negrini, R.; Souza Carneiro, P.L.; Bozzi, R. Population Structure and Genetic Diversity of Italian Beef Breeds as a Tool for Planning Conservation and Selection Strategies. Animals 2019, 9, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatkin, M. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Lewontin, R.C. The Interaction of Selection and Linkage. I. General Considerations; Heterotic Models. Genetics 1964, 49, 49–67. [Google Scholar]
- Park, L. Population-specific long-range linkage disequilibrium in the human genome and its influence on identifying common disease variants. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kruglyak, L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 1999, 22, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Cho, Y.M.; Lim, D.; Kim, H.C.; Choi, B.H.; Park, H.S.; Kim, O.H.; Kim, S.; Kim, T.H.; Yoon, D.; et al. Linkage Disequilibrium and Effective Population Size in Hanwoo Korean Cattle. Asian-Australas. J. Anim. Sci. 2011, 24, 1660–1665. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Goddard, M.E. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 2000, 155, 421–430. [Google Scholar] [PubMed]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Vander Voort, G.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front. Genet. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, M.; Bozzi, R.; García-Casco, J.; Núñez, Y.; Ribani, A.; Franci, O.; García, F.; Škrlep, M.; Schiavo, G.; Bovo, S.; et al. Genomic diversity, linkage disequilibrium and selection signatures in European local pig breeds assessed with a high density SNP chip. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Du, F.-X.; Clutter, A.C.; Lohuis, M.M. Characterizing Linkage Disequilibrium in Pig Populations. Int. J. Biol. Sci. 2007, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Lee, D.H.; Choi, N.; Sudrajad, P.; Lee, S.-H.; Lee, J.-H. Estimation of linkage disequilibrium and analysis of genetic diversity in Korean chicken lines. PLoS ONE 2018, 13, e0192063. [Google Scholar] [CrossRef] [Green Version]
- Ghoreishifar, S.M.; Moradi-Shahrbabak, H.; Parna, N.; Davoudi, P.; Khansefid, M. Linkage disequilibrium and within-breed genetic diversity in Iranian Zandi sheep. Arch. Tierz. 2019, 62, 143–151. [Google Scholar] [CrossRef]
- Monau, P.I.; Visser, C.; Muchadeyi, F.C.; Okpeku, M.; Nsoso, S.J.; Van Marle-Köster, E. Population structure of indigenous southern African goats based on the Illumina Goat50K SNP panel. Trop. Anim. Health Prod. 2020. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Dessie, T.; Lee, S.-H.; Kim, K.-S. Genome-wide linkage disequilibrium analysis of indigenous cattle breeds of Ethiopia and Korea using different SNP genotyping BeadChips. Genes Genom. 2015, 37, 759–765. [Google Scholar] [CrossRef]
- Jemaa, S.B.; Thamri, N.; Mnara, S.; Rebours, E.; Rocha, D.; Boussaha, M.; Jemaa, S.B.; Thamri, N.; Mnara, S.; Rebours, E.; et al. Linkage disequilibrium and past effective population size in native Tunisian cattle. Genet. Mol. Biol. 2019, 42, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Cañas-Álvarez, J.J.; Mouresan, E.F.; Varona, L.; Díaz, C.; Molina, A.; Baro, J.A.; Altarriba, J.; Carabaño, M.J.; Casellas, J.; Piedrafita, J. Linkage disequilibrium, persistence of phase, and effective population size in Spanish local beef cattle breeds assessed through a high-density single nucleotide polymorphism chip. J. Anim. Sci. 2016, 94, 2779–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef]
- Gautier, M.; Faraut, T.; Moazami-Goudarzi, K.; Navratil, V.; Foglio, M.; Grohs, C.; Boland, A.; Garnier, J.-G.; Boichard, D.; Lathrop, G.M.; et al. Genetic and Haplotypic Structure in 14 European and African Cattle Breeds. Genetics 2007, 177, 1059–1070. [Google Scholar] [CrossRef]
- Arias, J.A.; Keehan, M.; Fisher, P.; Coppieters, W.; Spelman, R. A high density linkage map of the bovine genome. BMC Genet. 2009, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J. Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1395–1409. [Google Scholar] [CrossRef] [Green Version]
- Corbin, L.J.; Liu, A.Y.H.; Bishop, S.C.; Woolliams, J.A. Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 2012, 129, 257–270. [Google Scholar] [CrossRef]
- Statistics of the Italian Limousine and Charolaise Breeds, ANACLI Site. Available online: http://www.anacli.it/WEBSITE/index.php?&pagid=2499&sessione= (accessed on 12 February 2020).
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 31 January 2020).
- Zhao, H.; Nettleton, D.; Dekkers, J.C.M. Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between single nucleotide polymorphisms. Genet. Res. 2007, 89, 1–6. [Google Scholar] [CrossRef]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Szyndler-Nędza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Bugno-Poniewierska, M. Comparison of linkage disequilibrium, effective population size and haplotype blocks in Polish Landrace and Polish native pig populations. Livest. Sci. 2020, 231, 103887. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Kukučková, V.; Moravčíková, N.; Ferenčaković, M.; Simčič, M.; Mészáros, G.; Sölkner, J.; Trakovická, A.; Kadlečík, O.; Curik, I.; Kasarda, R. Genomic characterization of Pinzgau cattle: Genetic conservation and breeding perspectives. Conserv. Genet. 2017, 18, 893–910. [Google Scholar] [CrossRef]
- Hayes, B.J. Novel Multilocus Measure of Linkage Disequilibrium to Estimate Past Effective Population Size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Ohta, T.; Kimura, M. Linkage Disequilibrium between Two Segregating Nucleotide Sites under the Steady Flux of Mutations in a Finite Population. Genetics 1971, 68, 571–580. [Google Scholar]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Qanbari, S. On the Extent of Linkage Disequilibrium in the Genome of Farm Animals. Front. Genet. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, M.; Eriksson, S.; Mikko, S.; Strandberg, E.; Stålhammar, H.; Groenen, M.A.M.; Crooijmans, R.P.M.A.; Andersson, G.; Johansson, A.M. Genomic relatedness and diversity of Swedish native cattle breeds. Genet. Sel. Evol. 2019, 51, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, A.R. How Population Growth Affects Linkage Disequilibrium. Genetics 2014, 197, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Pérez O’Brien, A.M.; Mészáros, G.; Utsunomiya, Y.T.; Sonstegard, T.S.; Garcia, J.F.; Van Tassell, C.P.; Carvalheiro, R.; da Silva, M.V.B.; Sölkner, J. Linkage disequilibrium levels in Bos indicus and Bos taurus cattle using medium and high density SNP chip data and different minor allele frequency distributions. Livest. Sci. 2014, 166, 121–132. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Saura, M.; Tolone, M.; Salces-Ortiz, J.; Di Gerlando, R.; Bertolini, F.; Fontanesi, L.; Sardina, M.T.; Serrano, M.; Portolano, B. The genome-wide structure of two economically important indigenous Sicilian cattle breeds1. J. Anim. Sci. 2014, 92, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, H.; Ahmad, N.; Heather, H.J.; Eui-soo, K.; Khan, W.A.; Ajmal, A.; Javed, K.; Pasha, T.N.; Ali, A.; Kim, J.J.; et al. Whole genome study of linkage disequilibrium in Sahiwal cattle. S. Afr. J. Anim. Sci. 2018, 48, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Makina, S.O.; Taylor, J.F.; van Marle-Köster, E.; Muchadeyi, F.C.; Makgahlela, M.L.; MacNeil, M.D.; Maiwashe, A. Extent of Linkage Disequilibrium and Effective Population Size in Four South African Sanga Cattle Breeds. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Zhu, B.; Wang, Y.H.; Wu, Y.; Xu, L.; Guo, L.P.; Yuan, Z.R.; Zhang, L.P.; Gao, X.; Gao, H.J.; et al. Linkage Disequilibrium Estimation of Chinese Beef Simmental Cattle Using High-density SNP Panels. Asian-Australas. J. Anim. Sci. 2013, 26, 772–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biegelmeyer, P.; Gulias-Gomes, C.C.; Caetano, A.R.; Steibel, J.P.; Cardoso, F.F. Linkage disequilibrium, persistence of phase and effective population size estimates in Hereford and Braford cattle. BMC Genet. 2016, 17, 32. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ramilo, S.T.; García-Cortés, L.A.; de Cara, M.Á.R. Artificial selection with traditional or genomic relationships: Consequences in coancestry and genetic diversity. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Khatkar, M.S.; Nicholas, F.W.; Collins, A.R.; Zenger, K.R.; Cavanagh, J.A.L.; Barris, W.; Schnabel, R.D.; Taylor, J.F.; Raadsma, H.W. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genom. 2008, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Lim, D.; Chai, H.-H.; Choi, B.-H.; Cho, Y. Demographic Trends in Korean Native Cattle Explained Using Bovine SNP50 Beadchip. Genom. Inform. 2016, 14, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Woolliams, J.A.; Gwaze, D.P.; Meuwissen, T.H.E.; Planchenault, D.; Renard, J.-P.; Thibier, M.; Wagner, H. Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans–Measurement of Domestic Animal Diversity (MoDAD); FAO: Rome, Italy, 1998; p. 58. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed 1 | N SNPs Pre-QC | N SNPs Post-QC | N Individuals Pre-QC | N Individuals Post-QC |
|---|---|---|---|---|
| CAL | 28,289 | 23,646 | 174 | 164 |
| MUP | 28,289 | 23,436 | 270 | 263 |
| PON | 28,289 | 22,791 | 44 | 41 |
| LIM | 28,289 | 23,279 | 100 | 100 |
| Breed 1 | CAL | MUP | PON | LIM | ||||
|---|---|---|---|---|---|---|---|---|
| Autosome | Average r2 | SD | Average r2 | SD | Average r2 | SD | Average r2 | SD |
| BTA1 | 0.14 | 0.20 | 0.13 | 0.20 | 0.20 | 0.25 | 0.08 | 0.16 |
| BTA2 | 0.16 | 0.23 | 0.13 | 0.19 | 0.17 | 0.22 | 0.10 | 0.18 |
| BTA3 | 0.15 | 0.21 | 0.12 | 0.17 | 0.16 | 0.22 | 0.08 | 0.15 |
| BTA4 | 0.14 | 0.19 | 0.17 | 0.26 | 0.20 | 0.26 | 0.10 | 0.22 |
| BTA5 | 0.15 | 0.20 | 0.15 | 0.20 | 0.18 | 0.23 | 0.07 | 0.12 |
| BTA6 | 0.18 | 0.26 | 0.14 | 0.20 | 0.19 | 0.23 | 0.08 | 0.14 |
| BTA7 | 0.12 | 0.16 | 0.13 | 0.17 | 0.17 | 0.21 | 0.07 | 0.12 |
| BTA8 | 0.14 | 0.19 | 0.11 | 0.16 | 0.16 | 0.21 | 0.06 | 0.11 |
| BTA9 | 0.15 | 0.22 | 0.17 | 0.23 | 0.15 | 0.21 | 0.08 | 0.17 |
| BTA10 | 0.13 | 0.18 | 0.13 | 0.17 | 0.15 | 0.18 | 0.06 | 0.10 |
| BTA11 | 0.12 | 0.18 | 0.12 | 0.16 | 0.15 | 0.18 | 0.06 | 0.11 |
| BTA12 | 0.12 | 0.17 | 0.14 | 0.19 | 0.17 | 0.21 | 0.06 | 0.10 |
| BTA13 | 0.15 | 0.21 | 0.12 | 0.17 | 0.16 | 0.20 | 0.05 | 0.09 |
| BTA14 | 0.13 | 0.18 | 0.13 | 0.18 | 0.19 | 0.23 | 0.06 | 0.11 |
| BTA15 | 0.14 | 0.18 | 0.15 | 0.21 | 0.20 | 0.24 | 0.09 | 0.17 |
| BTA16 | 0.15 | 0.22 | 0.15 | 0.22 | 0.23 | 0.28 | 0.12 | 0.20 |
| BTA17 | 0.13 | 0.18 | 0.15 | 0.20 | 0.15 | 0.18 | 0.06 | 0.10 |
| BTA18 | 0.13 | 0.17 | 0.15 | 0.20 | 0.17 | 0.20 | 0.06 | 0.12 |
| BTA19 | 0.15 | 0.20 | 0.14 | 0.19 | 0.16 | 0.21 | 0.07 | 0.13 |
| BTA20 | 0.21 | 0.27 | 0.15 | 0.21 | 0.20 | 0.25 | 0.13 | 0.21 |
| BTA21 | 0.14 | 0.21 | 0.19 | 0.24 | 0.22 | 0.25 | 0.10 | 0.18 |
| BTA22 | 0.12 | 0.16 | 0.11 | 0.16 | 0.14 | 0.17 | 0.05 | 0.08 |
| BTA23 | 0.14 | 0.20 | 0.14 | 0.19 | 0.15 | 0.19 | 0.08 | 0.15 |
| BTA24 | 0.14 | 0.19 | 0.12 | 0.16 | 0.17 | 0.22 | 0.06 | 0.11 |
| BTA25 | 0.13 | 0.17 | 0.12 | 0.16 | 0.15 | 0.19 | 0.06 | 0.10 |
| BTA26 | 0.13 | 0.17 | 0.13 | 0.18 | 0.18 | 0.22 | 0.05 | 0.09 |
| BTA27 | 0.11 | 0.15 | 0.10 | 0.14 | 0.15 | 0.18 | 0.05 | 0.09 |
| BTA28 | 0.12 | 0.16 | 0.13 | 0.16 | 0.15 | 0.19 | 0.06 | 0.10 |
| BTA29 | 0.15 | 0.22 | 0.12 | 0.17 | 0.20 | 0.25 | 0.05 | 0.09 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbri, M.C.; Dadousis, C.; Bozzi, R. Estimation of Linkage Disequilibrium and Effective Population Size in Three Italian Autochthonous Beef Breeds. Animals 2020, 10, 1034. https://doi.org/10.3390/ani10061034
Fabbri MC, Dadousis C, Bozzi R. Estimation of Linkage Disequilibrium and Effective Population Size in Three Italian Autochthonous Beef Breeds. Animals. 2020; 10(6):1034. https://doi.org/10.3390/ani10061034
Chicago/Turabian StyleFabbri, Maria Chiara, Christos Dadousis, and Riccardo Bozzi. 2020. "Estimation of Linkage Disequilibrium and Effective Population Size in Three Italian Autochthonous Beef Breeds" Animals 10, no. 6: 1034. https://doi.org/10.3390/ani10061034