Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, Genotyping, and Data Quality Control
2.2. Selection Criteria for Runs of Homozygosity
2.3. Distribution of Runs of Homozygosity
2.4. Genomic Inbreeding Coefficients
2.5. Identification of Common Runs of Homozygosity
3. Results
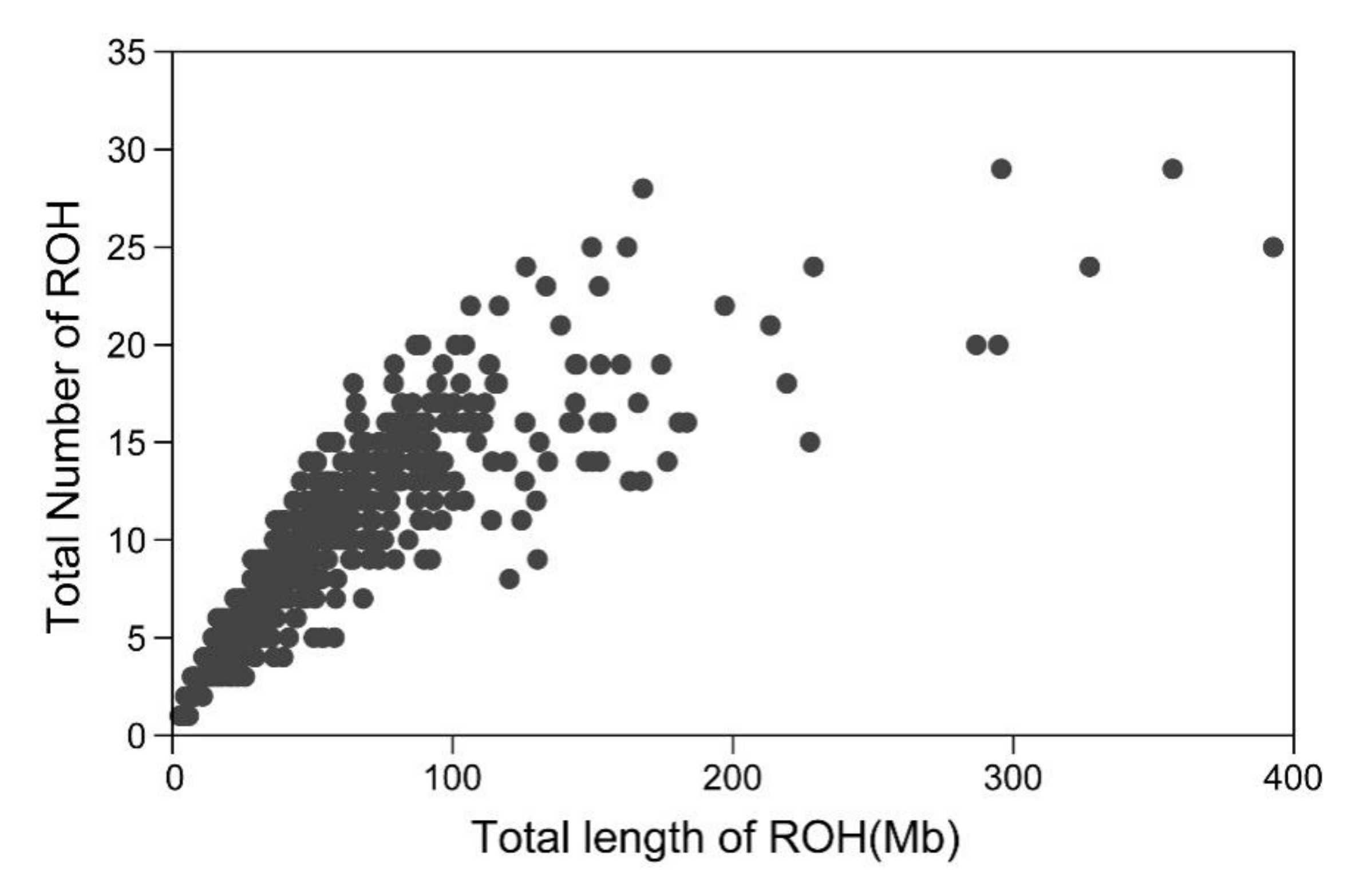
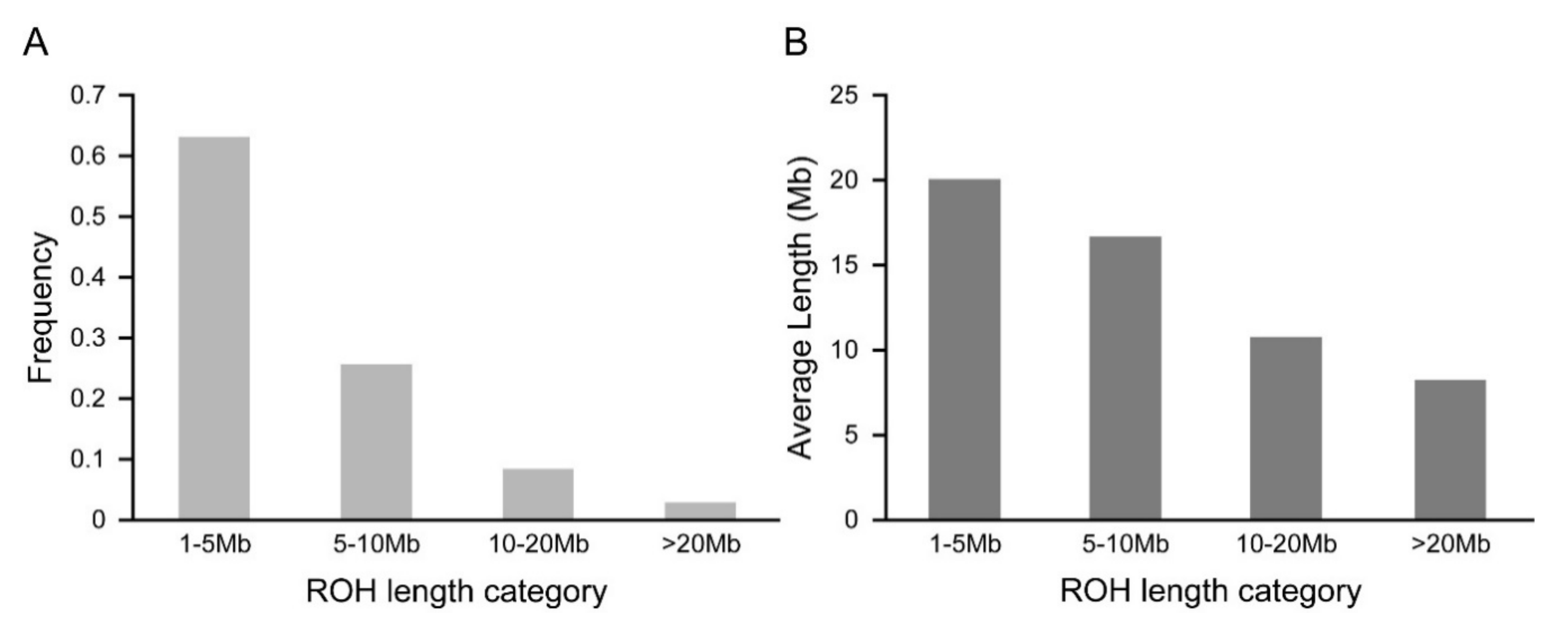
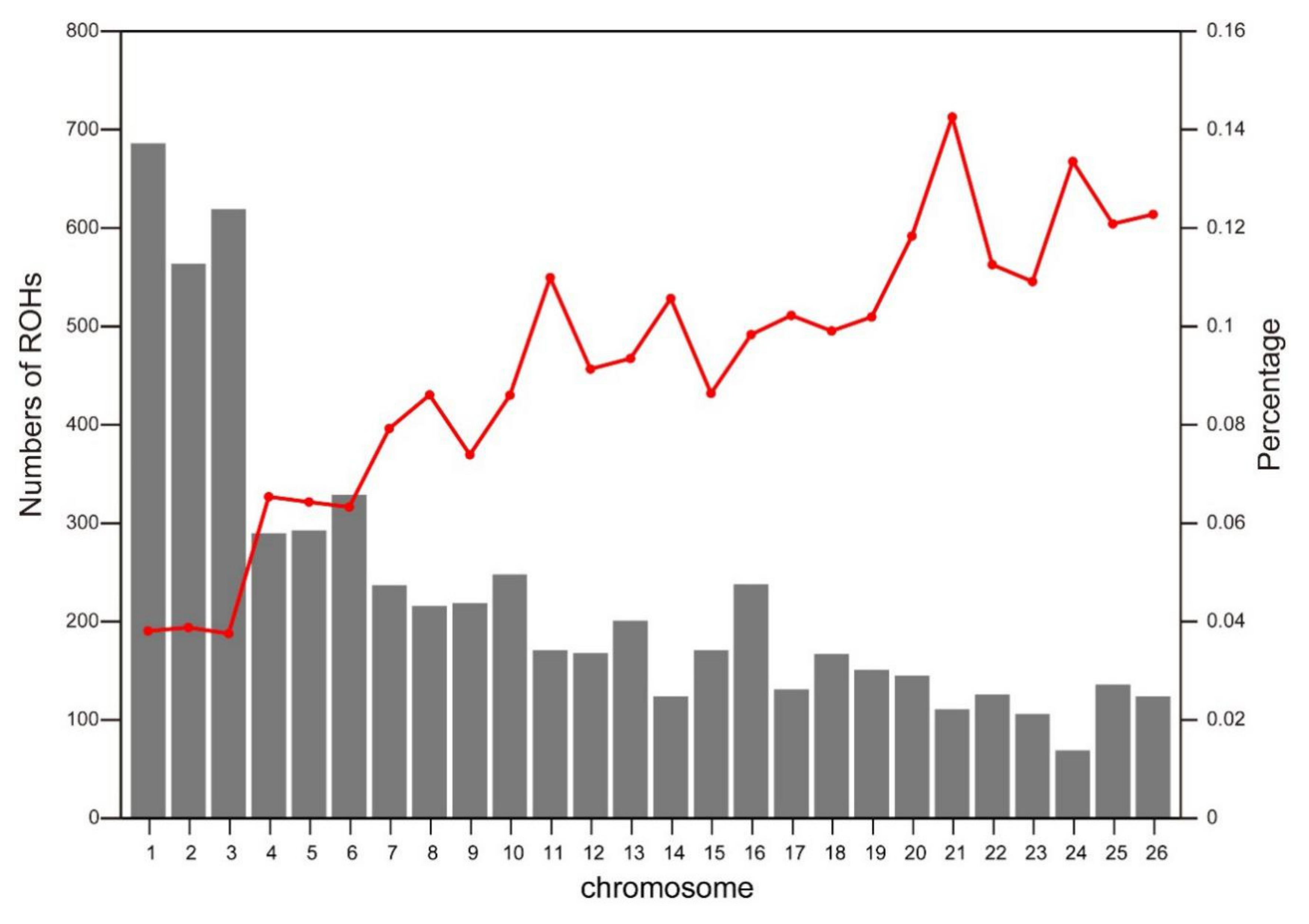
3.1. Distribution of Runs of Homozygosity
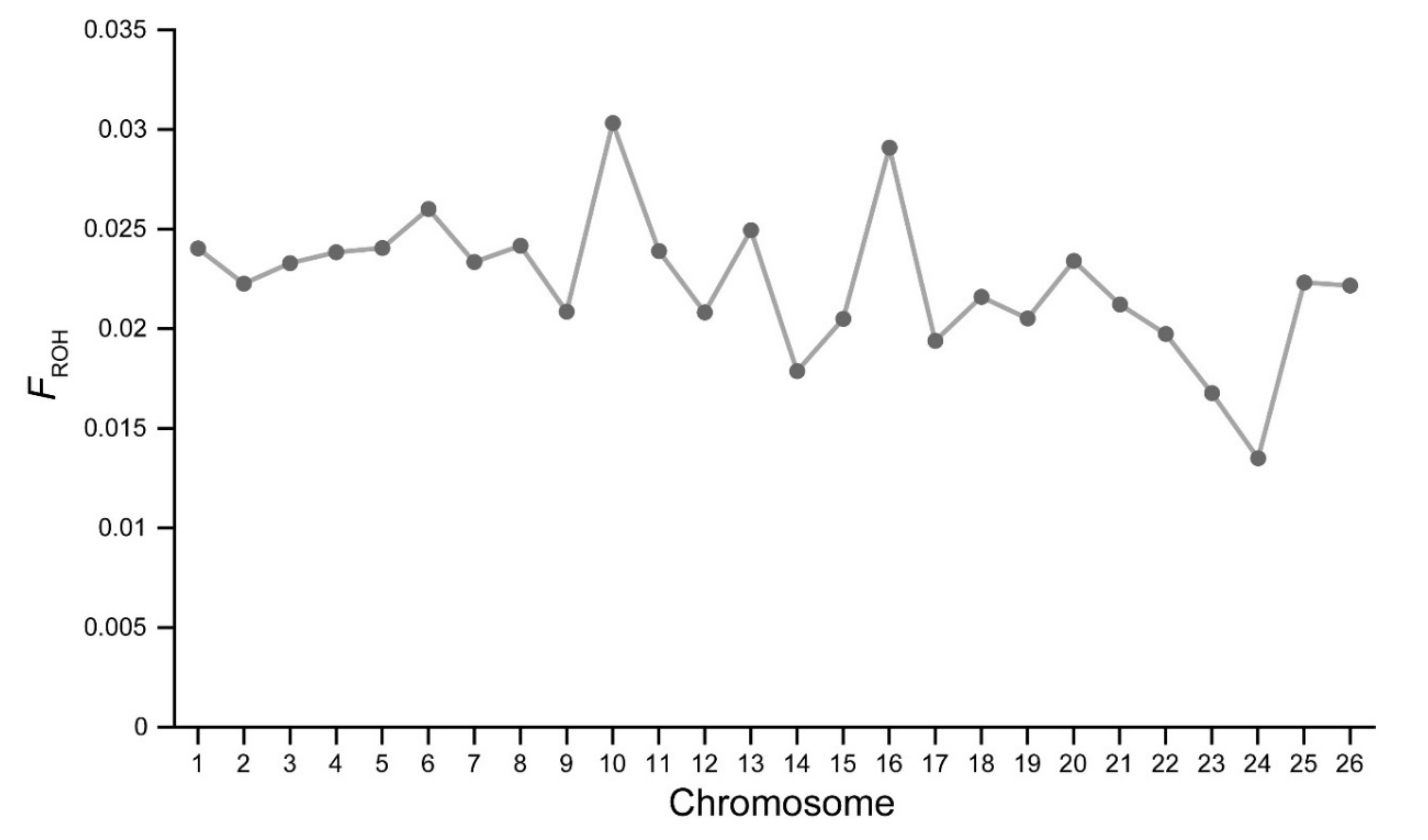
3.2. Inbreeding Coefficient Based on ROH
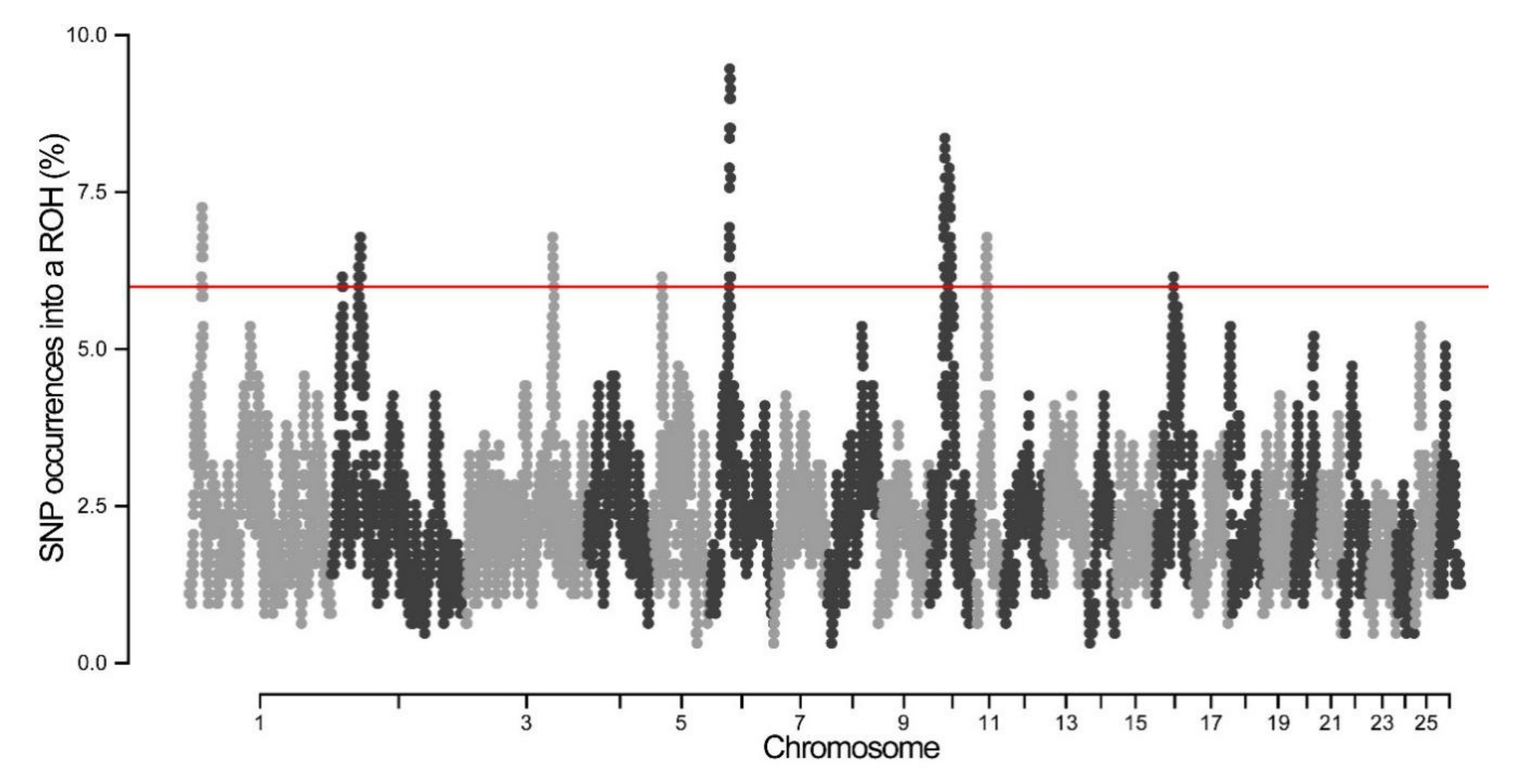
3.3. Genomic Regions with ROH Hotspots
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.; Megens, H.J.; Madsen, O.; Paudel, Y.; Frantz, L.A.F.; Schook, L.B.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Regions of Homozygosity in the Porcine Genome: Consequence of Demography and the Recombination Landscape. PLoS Genetics 2012, 8. [Google Scholar] [CrossRef] [Green Version]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Baumung, R.; Sölkner, J. Pedigree and marker information requirements to monitor genetic variability. Genet. Sel. Evol. 2003, 35, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Grilz-Seger, G.; Mesarič, M.; Cotman, M.; Neuditschko, M.; Druml, T.; Brem, G. Runs of homozygosity and population history of three horse breeds with small population size. J. Equine Vet. Sci. 2018, 71, 27–34. [Google Scholar] [CrossRef]
- Silió, L.; Rodríguez, M.; Fernández, A.; Barragán, C.; Benítez, R.; Óvilo, C.; Fernández, A. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J. Anim. Breed. Genet. 2013, 130, 349–360. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Chitneedi, P.; Arranz, J.; Suarez-Vega, A.; García-Gámez, E.; Gutiérrez-Gil, B. Estimations of linkage disequilibrium, effective population size and ROH-based inbreeding coefficients in Spanish Churra sheep using imputed high-density SNP genotypes. Anim. Genet. 2017, 48, 436–446. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.-S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangelo, S.; Sardina, M.; Tolone, M.; Di Gerlando, R.; Sutera, A.; Fontanesi, L.; Portolano, B. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds. Animal 2018, 12, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-S.; Sonstegard, T.S.; Rothschild, M.F. Recent artificial selection in US Jersey cattle impacts autozygosity levels of specific genomic regions. BMC Genom. 2015, 16, 302. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [Green Version]
- Signer-Hasler, H.; Burren, A.; Ammann, P.; Drögemüller, C.; Flury, C. Runs of homozygosity and signatures of selection: A comparison among eight local Swiss sheep breeds. Anim. Genet. 2019, 50, 512–525. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [Green Version]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Mesarič, M.; Cotman, M.; Brem, G. Analysis of ROH patterns in the Noriker horse breed reveals signatures of selection for coat color and body size. Anim. Genet. 2019, 50, 334–346. [Google Scholar] [CrossRef]
- Luigi-Sierra, M.; Cardoso, T.; Martínez, A.; Pons, A.; Bermejo, L.; Jordana, J.; Delgado, J.; Adán, S.; Ugarte, E.; Arranz, J. Low genome-wide homozygosity in 11 Spanish ovine breeds. Anim. Genet. 2019, 50, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.; Sottile, G.; Pilla, F.; Portolano, B.; Consortium, B.O.I. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.E.; Bérénos, C.; Slate, J.; Pemberton, J.M. Conserved genetic architecture underlying individual recombination rate variation in a wild population of Soay sheep (Ovis aries). Genetics 2016, 203, 583–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Al-Mamun, H.A.; Clark, S.A.; Kwan, P.; Gondro, C. Genome-wide linkage disequilibrium and genetic diversity in five populations of Australian domestic sheep. Genet. Sel. Evol. 2015, 47, 90. [Google Scholar] [CrossRef] [Green Version]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Neto, L.R.P.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; He, S.; Chen, L.; Li, W.; Di, J.; Liu, M. Estimates of linkage disequilibrium and effective population sizes in Chinese Merino (Xinjiang type) sheep by genome-wide SNPs. Genes Genom. 2017, 39, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef] [Green Version]
- Zavarez, L.B.; Utsunomiya, Y.T.; Carmo, A.S.; Neves, H.H.; Carvalheiro, R.; Ferenčaković, M.; Pérez, O.; Brien, A.M.; Curik, I.; Cole, J.B.; et al. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 2015, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Megdiche, S.; Mastrangelo, S.; Ben Hamouda, M.; Lenstra, J.A.; Ciani, E. Merino and Merino-derived sheep breeds: A further look at genome-wide selection signatures for wool traits. Front. Genet. 2019, 10, 1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fariello, M.-I.; Servin, B.; Tosser-Klopp, G.; Rupp, R.; Moreno, C.; San Cristobal, M.; Boitard, S.; Consortium, I.S.G. Selection signatures in worldwide sheep populations. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naval-Sanchez, M.; Nguyen, Q.; McWilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 2018, 9, 859. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Gil, B.; Esteban-Blanco, C.; Wiener, P.; Chitneedi, P.K.; Suarez-Vega, A.; Arranz, J.-J. High-resolution analysis of selection sweeps identified between fine-wool Merino and coarse-wool Churra sheep breeds. Genet. Sel. Evol. 2017, 49, 81. [Google Scholar] [CrossRef] [Green Version]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Walters, G.B.; Thorleifsson, G.; Stefansson, H.; Halldorsson, B.V.; Zusmanovich, P.; Sulem, P.; Thorlacius, S.; Gylfason, A.; Steinberg, S. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008, 40, 609. [Google Scholar] [CrossRef]
- Soranzo, N.; Rivadeneira, F.; Chinappen-Horsley, U.; Malkina, I.; Richards, J.B.; Hammond, N.; Stolk, L.; Nica, A.; Inouye, M.; Hofman, A. Meta-analysis of genome-wide scans for human adult stature identifies novel Loci and associations with measures of skeletal frame size. PLoS Genet. 2009, 5, e1000445. [Google Scholar] [CrossRef]
- Liu, R.; Sun, Y.; Zhao, G.; Wang, F.; Wu, D.; Zheng, M.; Chen, J.; Zhang, L.; Hu, Y.; Wen, J. Genome-wide association study identifies loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS ONE 2013, 8, e61172. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.K.; Sexten, A.K.; Kuehn, L.A.; Smith, T.P.; King, D.A.; Shackelford, S.D.; Wheeler, T.L.; Ferrell, C.L.; Jenkins, T.G.; Snelling, W.M. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011, 12, 103. [Google Scholar] [CrossRef] [Green Version]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.A. Four loci explain 83% of size variation in the horse. PLoS ONE 2012, 7, e39929. [Google Scholar] [CrossRef] [Green Version]
- Tetens, J.; Widmann, P.; Kühn, C.; Thaller, G. A genome-wide association study indicates LCORL/NCAPG as a candidate locus for withers height in G erman W armblood horses. Anim. Genet. 2013, 44, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-S.; Elbeltagy, A.; Aboul-Naga, A.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘guilt by association’-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef]
- Lv, F.-H.; Agha, S.; Kantanen, J.; Colli, L.; Stucki, S.; Kijas, J.W.; Joost, S.; Li, M.-H.; Ajmone Marsan, P. Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 2014, 31, 3324–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manunza, A.; Cardoso, T.F.; Noce, A.; Martínez, A.; Pons, A.; Bermejo, L.; Landi, V.; Sànchez, A.; Jordana, J.; Delgado, J.V. Population structure of eleven Spanish ovine breeds and detection of selective sweeps with BayeScan and hapFLK. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Li, S.; Liu, Q.; Wang, Z.; Zhou, Z.; Di, R.; Miao, B.; Hu, W.; Wang, X.; Hu, X. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 2018, 7, giy019. [Google Scholar] [CrossRef]
- Kardos, M.; Luikart, G.; Bunch, R.; Dewey, S.; Edwards, W.; McWilliam, S.; Stephenson, J.; Allendorf, F.W.; Hogg, J.T.; Kijas, J. Whole-genome resequencing uncovers molecular signatures of natural and sexual selection in wild bighorn sheep. Mol. Ecol. 2015, 24, 5616–5632. [Google Scholar] [CrossRef]
- Randhawa, I.A.S.; Khatkar, M.S.; Thomson, P.C.; Raadsma, H.W. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 2014, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.E.; Gratten, J.; Berenos, C.; Pilkington, J.G.; Clutton-Brock, T.H.; Pemberton, J.M.; Slate, J. Life history trade-offs at a single locus maintain sexually selected genetic variation. Nature 2013, 502, 93. [Google Scholar] [CrossRef]
- Dominik, S.; Henshall, J.; Hayes, B. A single nucleotide polymorphism on chromosome 10 is highly predictive for the polled phenotype in Australian Merino sheep. Anim. Genet. 2012, 43, 468–470. [Google Scholar] [CrossRef]
- Duijvesteijn, N.; Bolormaa, S.; Daetwyler, H.D.; van der Werf, J.H. Genomic prediction of the polled and horned phenotypes in Merino sheep. Genet. Sel. Evol. 2018, 50, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inbreeding Coefficient | Mean | Median | Minimum | Maximum | Coefficient of Variation | Number of Animals |
|---|---|---|---|---|---|---|
| FROH1-5Mb | 0.008 | 0.008 | 0.001 | 0.024 | 52.25 | 632 |
| FROH5-10Mb | 0.008 | 0.006 | 0.002 | 0.031 | 69.30 | 541 |
| FROH10-20Mb | 0.010 | 0.007 | 0.004 | 0.042 | 64.02 | 293 |
| FROH>20Mb | 0.022 | 0.014 | 0.008 | 0.109 | 88.24 | 99 |
| FROH>1Mb | 0.023 | 0.018 | 0.001 | 0.160 | 84.05 | 634 |
| No. | OAR | Start (bp) | Stop (bp) | Length (bp) | SNPs | Genes | cM/Mb |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 23294379 | 26901948 | 3607569 | 62 | 20 | 0.15 |
| 2 | 2 | 19930587 | 21080059 | 1149472 | 26 | 0 | 0.13 |
| 3 | 2 | 51106073 | 52409379 | 1303306 | 23 | 9 | 0.75 |
| 4 | 2 | 53415584 | 55758208 | 2342624 | 49 | 1 | 0.94 |
| 5 | 3 | 158391525 | 160963104 | 2571579 | 41 | 2 | 0.77 |
| 6 | 5 | 18659386 | 19857252 | 1197866 | 13 | 25 | 1.16 |
| 7 | 6 | 35075727 | 38668794 | 3593067 | 59 | 18 | 0.79 |
| 8 | 10 | 27305619 | 29842383 | 2536764 | 61 | 9 | 1.32 |
| 9 | 10 | 34325096 | 41802553 | 7477457 | 132 | 27 | 0.27 |
| 10 | 11 | 25425573 | 26939891 | 1514318 | 22 | 73 | 1.18 |
| 11 | 11 | 28019509 | 28518859 | 493350 | 14 | 5 | 0.96 |
| 12 | 11 | 28969704 | 28969704 | 0 | 1 | 1 | / |
| 13 | 16 | 32945561 | 32982579 | 37018 | 2 | 0 | 0 |
| Category | Description | −log10P |
|---|---|---|
| GO Biological Processes | ||
| GO:0000079 | Regulation of cyclin-dependent protein serine/threonine kinase activity | 3.84 |
| GO:0002639 | Positive regulation of immunoglobulin production | 3.47 |
| GO:0048599 | Oocyte development | 3.36 |
| GO:0048839 | Inner ear development | 3.12 |
| GO:0007568 | Aging | 3.03 |
| GO:0060079 | Excitatory postsynaptic potential | 2.96 |
| GO:0031214 | Biomineral tissue development | 2.76 |
| GO:0036465 | Synaptic vesicle recycling | 2.69 |
| GO:1902692 | Regulation of neuroblast proliferation | 2.68 |
| GO:0047496 | Vesicle transport along microtubule | 2.24 |
| GO:0043065 | Positive regulation of apoptotic process | 2.20 |
| GO:0051052 | Regulation of DNA metabolic process | 2.20 |
| GO:0040008 | Regulation of growth | 2.15 |
| GO:0007566 | Embryo implantation | 2.14 |
| Reactome Gene Sets | ||
| R-HSA-5099900 | WNT5A-dependent internalization of FZD4 | 3.71 |
| R-HSA-190828 | Gap junction trafficking | 3.32 |
| Canonical Pathways | ||
| M219 | PID hedgehog-GLI pathway | 3.28 |
| KEGG Pathway | ||
| hsa04918 | Thyroid hormone synthesis | 2.58 |
| hsa04961 | Endocrine and other factor-regulated calcium reabsorption | 2.24 |
| hsa00330 | Arginine and proline metabolism | 2.16 |
| OAR | Position (Mb) | Overlap Selection Signature Reference | Candidate Gene | Function |
|---|---|---|---|---|
| 2 | 51.10–52.41 | Lv Fenghua et al. [44] | MELK, GNE | Environment adaption |
| 5 | 18.66–19.86 | Naval-Sanchez et al. [33] | IL4, IL13 IL5, IRF1 | Immune function |
| 6 | 35.08–38.67 | Fariello et al. [32] Gutiérrez-Gil et al. [34] Naval-Sanchez et al. [33] Signer-Hasler et al. [18] | NCAPG, LCORL | Weight/height |
| 10 | 27.31–29.84 | Kijas et al. [27] Manunza et al. [45] Fariello et al. [32] Pan et al. [46] Kardos et al. [47] Randhawa et al. [48] | RXFP2 | Horn |
| 11 | 28.02–28.52 | Signer-Hasler et al. [18] Kim et al. [42] | FGF11, TP53 | Body size |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, S.; Di, J.; Han, B.; Chen, L.; Liu, M.; Li, W. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino. Animals 2020, 10, 524. https://doi.org/10.3390/ani10030524
He S, Di J, Han B, Chen L, Liu M, Li W. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino. Animals. 2020; 10(3):524. https://doi.org/10.3390/ani10030524
Chicago/Turabian StyleHe, Sangang, Jiang Di, Bing Han, Lei Chen, Mingjun Liu, and Wenrong Li. 2020. "Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino" Animals 10, no. 3: 524. https://doi.org/10.3390/ani10030524