
Methods for the Manipulation of Herpesvirus Genome and the Application to Marek’s Disease Virus Research


Abstract
:1. Introduction
2. Temperature Sensitive (ts) Mutant
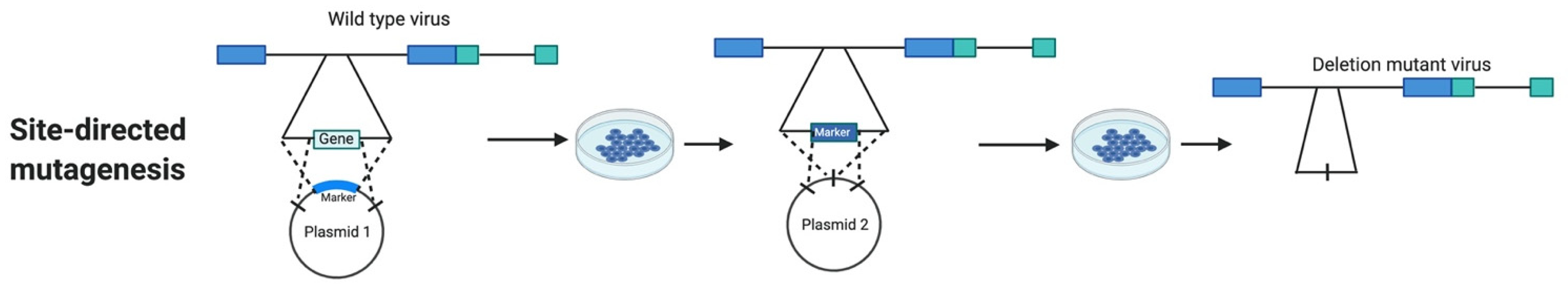
3. Marker Assisted Site-Directed Mutagenesis
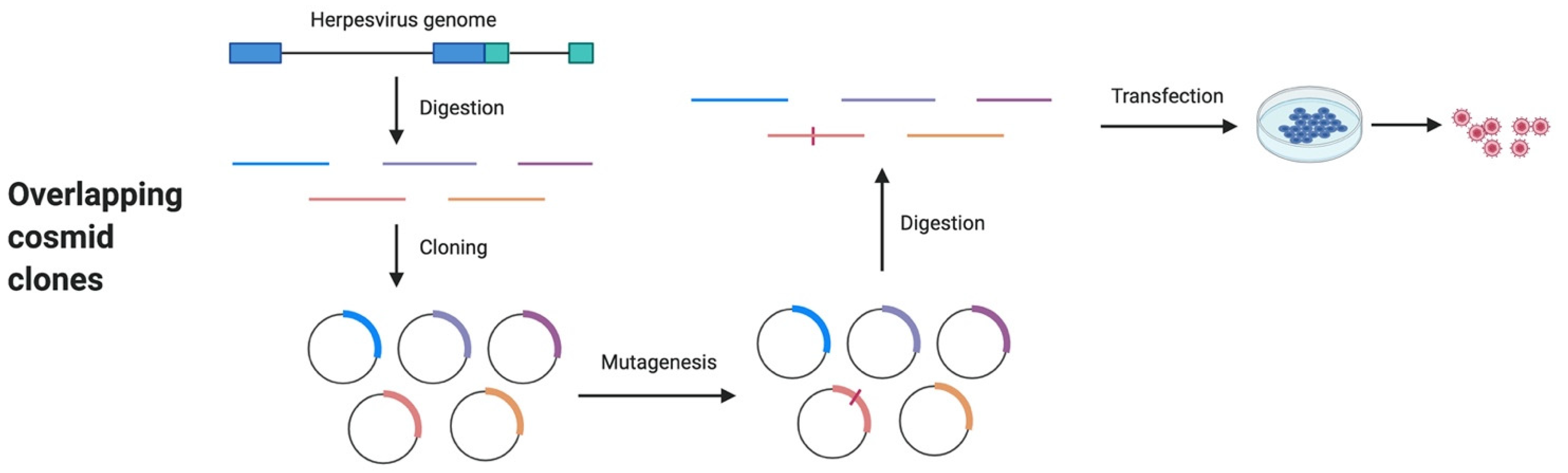
4. Overlapping Cosmid Clones
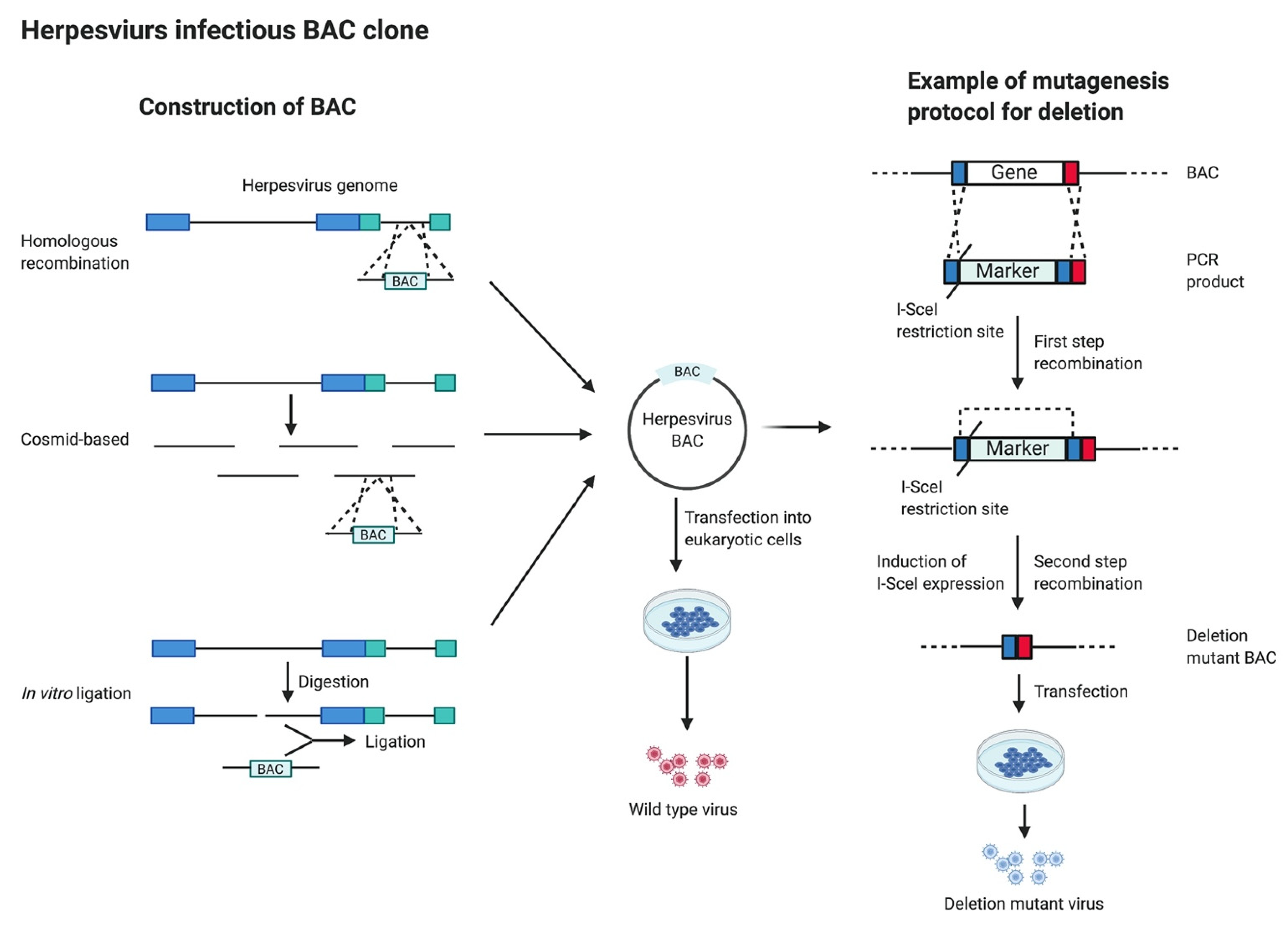
5. Infectious Bacterial Artificial Chromosome (BAC) Clones
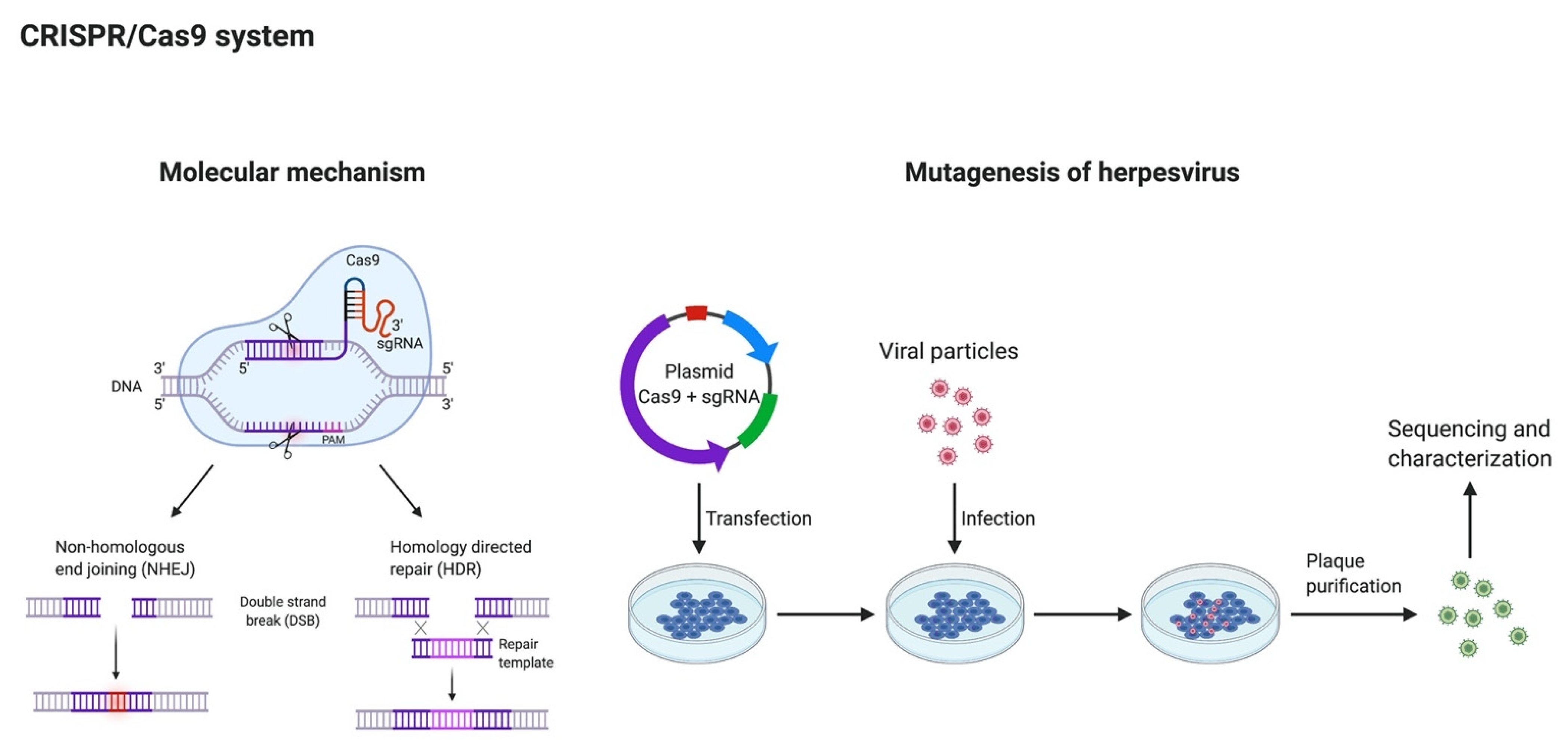
6. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 System
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Payne, S. (Ed.) Family Herpesviridae. In Viruses: From Understanding to Investigation; Elsevier: Amsterdam, The Netherlands, 2017; pp. 269–278. [Google Scholar] [CrossRef]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral. Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [Green Version]
- Marek, J. Multiple Nerventzuendung (Polyneuritis) bei Huehnern. Dtsch. Tierarztl. Wochenschr. 1907, 15, 417–421. [Google Scholar]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s disease virus: From miasma to model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Churchill, A.E.; Payne, L.N.; Chubb, R.C. Immunization against Marek’s disease using a live attenuated virus. Nature 1969, 221, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Izumiya, Y.; Lupiani, B. Marek’s disease vaccines: Current status, and strategies for improvement and development of vector vaccines. Vet. Microbiol. 2017, 206, 113–120. [Google Scholar] [CrossRef]
- Okazaki, W.; Purchase, H.G.; Burmester, B.R. Protection against Marek’s disease by vaccination with a herpesvirus of turkeys. Avian Dis. 1970, 14, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Schat, K.A. History of the First-Generation Marek’s Disease Vaccines: The Science and Little-Known Facts. Avian Dis. 2016, 60, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Rispens, B.H.; van Vloten, H.; Mastenbroek, N.; Maas, H.J.; Schat, K.A. Control of Marek’s disease in the Netherlands. I. Isolation of an avirulent Marek’s disease virus (strain CVI 988) and its use in laboratory vaccination trials. Avian Dis. 1972, 16, 108–125. [Google Scholar] [CrossRef]
- Witter, R.L. Protection by attenuated and polyvalent vaccines against highly virulent strains of Marek’s disease virus. Avian Pathol. 1982, 11, 49–62. [Google Scholar] [CrossRef]
- Witter, R.L.; Lee, L.F. Polyvalent Marek’s disease vaccines: Safety, efficacy and protective synergism in chickens with maternal antibodies. Avian Pathol. 1984, 13, 75–92. [Google Scholar] [CrossRef] [Green Version]
- Davison, F.; Nair, V. Use of Marek’s disease vaccines: Could they be driving the virus to increasing virulence? Expert Rev. Vaccines 2005, 4, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, B.B.; Arndt, S.; Trapp, S.; Osterrieder, N.; Jarosinski, K.W. Herpesvirus telomerase RNA (vTR) with a mutated template sequence abrogates herpesvirus-induced lymphomagenesis. PLoS Pathog. 2011, 7, e1002333. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Lupiani, B.; Silva, R.F.; Kung, H.J.; Reddy, S.M. Recombinant Marek’s disease virus (MDV) lacking the Meq oncogene confers protection against challenge with a very virulent plus strain of MDV. Vaccine 2008, 26, 1887–1892. [Google Scholar] [CrossRef]
- Trapp, S.; Parcells, M.S.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Kumar, P.M.; Nair, V.K.; Osterrieder, N. A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J. Exp. Med. 2006, 203, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Lupiani, B.; Lee, L.F.; Cui, X.; Gimeno, I.; Anderson, A.; Morgan, R.W.; Silva, R.F.; Witter, R.L.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 2004, 101, 11815–11820. [Google Scholar] [CrossRef] [Green Version]
- Burge, B.W.; Pfefferkorn, E.R. Conditional Lethal Mutants of an Rna Animal Virus. Virology 1964, 24, 126–128. [Google Scholar] [CrossRef]
- Burge, B.W.; Pfefferkorn, E.R. Conditional-Lethal Mutants of an Animal Virus: Identification of Two Cistrons. Science 1965, 148, 959–960. [Google Scholar] [CrossRef]
- Fenner, F. Conditional lethal mutants of animal viruses. Curr. Top. Microbiol. Immunol. 1969, 48, 1–28. [Google Scholar] [CrossRef]
- Schaffer, P.; Vonka, V.; Lewis, R.; Benyesh-Melnick, M. Temperature-sensitive mutants of herpes simplex virus. Virology 1970, 42, 1144–1146. [Google Scholar] [CrossRef]
- Schaffer, P.A.; Courtney, R.J.; McCombs, R.M.; Benyesh-Melnick, M. A temperature-sensitive mutant of herpes simplex virus defective in glycoprotein synthesis. Virology 1971, 46, 356–368. [Google Scholar] [CrossRef]
- Schaffer, P.A.; Aron, G.M.; Biswal, N.; Benyesh-Melnick, M. Temperature-sensitive mutants of herpes simplex virus type 1: Isolation, complementation and partial characterization. Virology 1973, 52, 57–71. [Google Scholar] [CrossRef]
- Schaffer, P.A.; Carter, V.C.; Timbury, M.C. Collaborative complementation study of temperature-sensitive mutants of herpes simplex virus types 1 and 2. J. Virol. 1978, 27, 490–504. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, P.A.; Weller, S.K.; Pancake, B.A.; Coen, D.M. Genetics of herpes simplex virus. J. Investig. Dermatol. 1984, 83, 42s–47s. [Google Scholar] [CrossRef] [Green Version]
- Feldman, L.; Blankenship, M.L.; Ben-Porat, T. Isolation and characterization of a temperature-sensitive uncoating mutant of pseudorabies virus. J. Gen. Virol. 1981, 54, 333–342. [Google Scholar] [CrossRef]
- Shiraki, K.; Ogino, T.; Yamanishi, K.; Takahashi, M. Isolation of drug resistant mutants of varicella-zoster virus: Cross resistance of acyclovir resistant mutants with phosphonoacetic acid and bromodeoxyuridine. Biken J. 1983, 26, 17–23. [Google Scholar] [PubMed]
- Akel, H.M.; Sweet, C. Isolation and preliminary characterisation of twenty-five temperature-sensitive mutants of mouse cytomegalovirus. FEMS Microbiol. Lett. 1993, 113, 253–260. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, R.T.; Summers, W.C. Isolation and characterization of phosphonoacetic acid-resistant mutants of human cytomegalovirus. J. Virol. 1987, 61, 1291–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witter, R.L.; Offenbecker, L. Nonprotective and temperature-sensitive variants of Marek’s disease vaccine viruses. J. Natl. Cancer Inst. 1979, 62, 143–151. [Google Scholar] [PubMed]
- Wigler, M.; Silverstein, S.; Lee, L.S.; Pellicer, A.; Cheng, Y.; Axel, R. Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell 1977, 11, 223–232. [Google Scholar] [CrossRef]
- Pellicer, A.; Wigler, M.; Axel, R.; Silverstein, S. The transfer and stable integration of the HSV thymidine kinase gene into mouse cells. Cell 1978, 14, 133–141. [Google Scholar] [CrossRef]
- Enquist, L.W.; Vande Woude, G.F.; Wagner, M.; Smiley, J.R.; Summers, W.C. Construction and characterization of a recombinant plasmid encoding the gene for the thymidine kinase of Herpes simplex type 1 virus. Gene 1979, 7, 335–342. [Google Scholar] [CrossRef]
- Smiley, J.R. Construction in vitro and rescue of a thymidine kinase-deficient deletion mutation of herpes simplex virus. Nature 1980, 285, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Post, L.E.; Roizman, B. A generalized technique for deletion of specific genes in large genomes: Alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 1981, 25, 227–232. [Google Scholar] [CrossRef]
- Parcells, M.S.; Anderson, A.S.; Morgan, T.W. Retention of oncogenicity by a Marek’s disease virus mutant lacking six unique short region genes. J. Virol. 1995, 69, 7888–7898. [Google Scholar] [CrossRef] [Green Version]
- Parcells, M.S.; Lin, S.F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): Characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.M.; Lupiani, B.; Gimeno, I.M.; Silva, R.F.; Lee, L.F.; Witter, R.L. Rescue of a pathogenic Marek’s disease virus with overlapping cosmid DNAs: Use of a pp38 mutant to validate the technology for the study of gene function. Proc. Natl. Acad. Sci. USA 2002, 99, 7054–7059. [Google Scholar] [CrossRef] [Green Version]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Reddy, S.M.; Lee, L.F.; Silva, R.F. The pp38 gene of Marek’s disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J. Virol. 2005, 79, 4545–4549. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Lee, L.F.; Reed, W.M.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded vIL-8 gene is involved in early cytolytic infection but dispensable for establishment of latency. J. Virol. 2004, 78, 4753–4760. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Lee, L.F.; Hunt, H.D.; Reed, W.M.; Lupiani, B.; Reddy, S.M. A Marek’s disease virus vIL-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 2005, 49, 199–206. [Google Scholar] [CrossRef]
- Suchodolski, P.F.; Izumiya, Y.; Lupiani, B.; Ajithdoss, D.K.; Gilad, O.; Lee, L.F.; Kung, H.J.; Reddy, S.M. Homodimerization of Marek’s disease virus-encoded Meq protein is not sufficient for transformation of lymphocytes in chickens. J. Virol. 2009, 83, 859–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchodolski, P.F.; Izumiya, Y.; Lupiani, B.; Ajithdoss, D.K.; Lee, L.F.; Kung, H.J.; Reddy, S.M. Both homo and heterodimers of Marek’s disease virus encoded Meq protein contribute to transformation of lymphocytes in chickens. Virology 2010, 399, 312–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.F.; Silva, R.F.; Cui, X.; Zhang, H.; Heidari, M.; Reddy, S.M. Characterization of LORF11, a unique gene common to the three Marek’s disease virus serotypes. Avian Dis. 2007, 51, 851–857. [Google Scholar] [CrossRef]
- Schumacher, D.; Tischer, B.K.; Fuchs, W.; Osterrieder, N. Reconstitution of Marek’s disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J. Virol. 2000, 74, 11088–11098. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.C.; Baigent, S.J.; Smith, L.P.; Chattoo, J.P.; Petherbridge, L.J.; Hawes, P.; Allday, M.J.; Nair, V. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek’s disease virus. Proc. Natl. Acad. Sci. USA 2006, 103, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xu, H.; Yao, Y.; Smith, L.P.; Kgosana, L.; Green, J.; Petherbridge, L.; Baigent, S.J.; Nair, V. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog. 2011, 7, e1001305. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Margulis, N.G.; Kamil, J.P.; Spatz, S.J.; Nair, V.K.; Osterrieder, N. Horizontal transmission of Marek’s disease virus requires US2, the UL13 protein kinase, and gC. J. Virol. 2007, 81, 10575–10587. [Google Scholar] [CrossRef] [Green Version]
- Krieter, A.; Ponnuraj, N.; Jarosinski, K.W. Expression of the Conserved Herpesvirus Protein Kinase (CHPK) of Marek’s Disease Alphaherpesvirus in the Skin Reveals a Mechanistic Importance for CHPK during Interindividual Spread in Chickens. J. Virol. 2020, 94, e01522-19. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Osterrieder, N. Further analysis of Marek’s disease virus horizontal transmission confirms that U(L)44 (gC) and U(L)13 protein kinase activity are essential, while U(S)2 is nonessential. J. Virol. 2010, 84, 7911–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosinski, K.W.; Osterrieder, N. Marek’s disease virus expresses multiple UL44 (gC) variants through mRNA splicing that are all required for efficient horizontal transmission. J. Virol. 2012, 86, 7896–7906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuard, A.; Courvoisier-Guyader, K.; Remy, S.; Spatz, S.; Denesvre, C.; Pasdeloup, D. The Tegument Protein pUL47 of Marek’s Disease Virus Is Necessary for Horizontal Transmission and Is Important for Expression of Glycoprotein gC. J. Virol. 2020, 95, e01645-20. [Google Scholar] [CrossRef] [PubMed]
- Ponnuraj, N.; Tien, Y.T.; Vega-Rodriguez, W.; Krieter, A.; Jarosinski, K.W. The Herpesviridae Conserved Multifunctional Infected-Cell Protein 27 (ICP27) Is Important but Not Required for Replication and Oncogenicity of Marek’s Disease Alphaherpesvirus. J. Virol. 2019, 93, e01903-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; Tischer, B.K.; Trapp, S.; Osterrieder, N. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 2005, 79, 3987–3997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; McKinney, C.; Kaufer, B.B.; Osterrieder, N. Enzymatically inactive U(S)3 protein kinase of Marek’s disease virus (MDV) is capable of depolymerizing F-actin but results in accumulation of virions in perinuclear invaginations and reduced virus growth. Virology 2008, 375, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Lupiani, B.; Bajwa, K.; Khan, O.A.; Izumiya, Y.; Reddy, S.M. Role of Marek’s Disease Virus (MDV)-Encoded US3 Serine/Threonine Protein Kinase in Regulating MDV Meq and Cellular CREB Phosphorylation. J. Virol. 2020, 94, e00892-20. [Google Scholar] [CrossRef]
- Liao, Y.; Lupiani, B.; Ai-Mahmood, M.; Reddy, S.M. Marek’s disease virus US3 protein kinase phosphorylates chicken HDAC 1 and 2 and regulates viral replication and pathogenesis. PLoS Pathog. 2021, 17, e1009307. [Google Scholar] [CrossRef]
- Dorange, F.; Tischer, B.K.; Vautherot, J.F.; Osterrieder, N. Characterization of Marek’s disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 2002, 76, 1959–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosinski, K.W.; Arndt, S.; Kaufer, B.B.; Osterrieder, N. Fluorescently tagged pUL47 of Marek’s disease virus reveals differential tissue expression of the tegument protein in vivo. J. Virol. 2012, 86, 2428–2436. [Google Scholar] [CrossRef] [Green Version]
- Tai, S.S.; Hearn, C.; Umthong, S.; Agafitei, O.; Cheng, H.H.; Dunn, J.R.; Niikura, M. Expression of Marek’s Disease Virus Oncoprotein Meq During Infection in the Natural Host. Virology 2017, 503, 103–113. [Google Scholar] [CrossRef]
- Remy, S.; Blondeau, C.; Le Vern, Y.; Lemesle, M.; Vautherot, J.F.; Denesvre, C. Fluorescent tagging of VP22 in N-terminus reveals that VP22 favors Marek’s disease virus (MDV) virulence in chickens and allows morphogenesis study in MD tumor cells. Vet. Res. 2013, 44, 125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tang, N.; Sadigh, Y.; Baigent, S.; Shen, Z.; Nair, V.; Yao, Y. Application of CRISPR/Cas9 Gene Editing System on MDV-1 Genome for the Study of Gene Function. Viruses 2018, 10, 279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Luo, J.; Tang, N.; Teng, M.; Reddy, V.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Targeted Editing of the pp38 Gene in Marek’s Disease Virus-Transformed Cell Lines Using CRISPR/Cas9 System. Viruses 2019, 11, 391. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tang, N.; Luo, J.; Teng, M.; Moffat, K.; Shen, Z.; Watson, M.; Nair, V.; Yao, Y. Marek’s Disease Virus-Encoded MicroRNA 155 Ortholog Critical for the Induction of Lymphomas Is Not Essential for the Proliferation of Transformed Cell Lines. J. Virol. 2019, 93, e00713-19. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Teng, M.; Zai, X.; Tang, N.; Zhang, Y.; Mandviwala, A.; Reddy, V.; Baigent, S.; Yao, Y.; Nair, V. Efficient Mutagenesis of Marek’s Disease Virus-Encoded microRNAs Using a CRISPR/Cas9-Based Gene Editing System. Viruses 2020, 12, 466. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.; Hohn, B. Cosmids: A type of plasmid gene-cloning vector that is packageable in vitro in bacteriophage lambda heads. Proc. Natl. Acad. Sci. USA 1978, 75, 4242–4246. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.; Bruning, H.J. Plasmids useable as gene-cloning vectors in an in vitro packaging by coliphage lambda: “Cosmids”. Gene 1978, 4, 85–107. [Google Scholar] [CrossRef]
- van Zijl, M.; Quint, W.; Briaire, J.; de Rover, T.; Gielkens, A.; Berns, A. Regeneration of herpesviruses from molecularly cloned subgenomic fragments. J. Virol. 1988, 62, 2191–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, C.; Davison, A.J. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 1993, 197, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Robertson, E.; Yalamanchili, R.; Longnecker, R.; Kieff, E. Epstein-Barr virus recombinants from overlapping cosmid fragments. J. Virol. 1993, 67, 7298–7306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.I.; Seidel, K.E. Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 7376–7380. [Google Scholar] [CrossRef] [Green Version]
- Nicolson, L.; Rafferty, E.L.; Brawley, A.; Onions, D.E. An improved cosmid vector for the cloning of equine herpesvirus DNA. Gene 1994, 150, 405–406. [Google Scholar] [CrossRef]
- Reilly, J.D.; Silva, R.F. Cosmid library of the turkey herpesvirus genome constructed from nanogram quantities of viral DNA associated with an excess of cellular DNA. J. Virol. Methods 1993, 41, 323–331. [Google Scholar] [CrossRef]
- Lindenmaier, W.; Bauer, H.J. Cosmid cloning and restriction endonuclease mapping of the herpesvirus of turkeys (HVT) genome. Arch. Virol. 1994, 135, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, B.C.; Hubinette, M.M.; Qiang, D.; MacDonald, M.L.; Tufaro, F. Allele replacement: An application that permits rapid manipulation of herpes simplex virus type 1 genomes. Gene Ther. 1999, 6, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, N. Bacteriophage P1 cloning system for the isolation, amplification, and recovery of DNA fragments as large as 100 kilobase pairs. Proc. Natl. Acad. Sci. USA 1990, 87, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, P.A.; Amemiya, C.T.; Garnes, J.; Kroisel, P.M.; Shizuya, H.; Chen, C.; Batzer, M.A.; de Jong, P.J. A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nat. Genet. 1994, 6, 84–89. [Google Scholar] [CrossRef]
- Shizuya, H.; Birren, B.; Kim, U.J.; Mancino, V.; Slepak, T.; Tachiiri, Y.; Simon, M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA 1992, 89, 8794–8797. [Google Scholar] [CrossRef] [Green Version]
- Luckow, V.A.; Lee, S.C.; Barry, G.F.; Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 1993, 67, 4566–4579. [Google Scholar] [CrossRef] [Green Version]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; Kaufer, B.B. Viral bacterial artificial chromosomes: Generation, mutagenesis, and removal of mini-F sequences. J. Biomed. Biotechnol. 2012, 2012, 472537. [Google Scholar] [CrossRef] [Green Version]
- Warden, C.; Tang, Q.; Zhu, H. Herpesvirus BACs: Past, present, and future. J. Biomed. Biotechnol. 2011, 2011, 124595. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Rowe, J.; Wang, W.; Sommer, M.; Arvin, A.; Moffat, J.; Zhu, H. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J. Virol. 2007, 81, 9024–9033. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; Kaufer, B.B.; Sommer, M.; Wussow, F.; Arvin, A.M.; Osterrieder, N. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 2007, 81, 13200–13208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niikura, M.; Kim, T.; Silva, R.F.; Dodgson, J.; Cheng, H.H. Virulent Marek’s disease virus generated from infectious bacterial artificial chromosome clones with complete DNA sequence and the implication of viral genetic homogeneity in pathogenesis. J. Gen. Virol. 2011, 92, 598–607. [Google Scholar] [CrossRef]
- Borenstein, R.; Frenkel, N. Cloning human herpes virus 6A genome into bacterial artificial chromosomes and study of DNA replication intermediates. Proc. Natl. Acad. Sci. USA 2009, 106, 19138–19143. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.N.; Meers, J.; Fowler, E.; Mahony, T. Back to BAC: The use of infectious clone technologies for viral mutagenesis. Viruses 2012, 4, 211–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, W.; Menard, C.; Hobom, U.; Odenbreit, S.; Messerle, M.; Koszinowski, U.H. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nat. Biotechnol. 1999, 17, 360–364. [Google Scholar] [CrossRef]
- Chattoo, J.P.; Stevens, M.P.; Nair, V. Rapid identification of non-essential genes for in vitro replication of Marek’s disease virus by random transposon mutagenesis. J. Virol. Methods 2006, 135, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Sollars, P.J.; Smith, G.A. New tools to convert bacterial artificial chromosomes to a self-excising design and their application to a herpes simplex virus type 1 infectious clone. BMC Biotechnol. 2016, 16, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef]
- Petherbridge, L.; Brown, A.C.; Baigent, S.J.; Howes, K.; Sacco, M.A.; Osterrieder, N.; Nair, V.K. Oncogenicity of virulent Marek’s disease virus cloned as bacterial artificial chromosomes. J. Virol. 2004, 78, 13376–13380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Wang, Y.; Shi, X.; Tong, G.; Lan, D.; He, L.; Qiu, H.; Liu, C.; Wang, M. Construction of Marek’s disease virus serotype 814 strain as an infectious bacterial artificial chromosome. Sheng Wu Gong Cheng Xue Bao 2008, 24, 569–575. [Google Scholar] [CrossRef]
- Reddy, S.M.; Sun, A.; Khan, O.A.; Lee, L.F.; Lupiani, B. Cloning of a very virulent plus, 686 strain of Marek’s disease virus as a bacterial artificial chromosome. Avian Dis. 2013, 57, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Petherbridge, L.; Xu, H.; Zhao, Y.; Smith, L.P.; Simpson, J.; Baigent, S.; Nair, V. Cloning of Gallid herpesvirus 3 (Marek’s disease virus serotype-2) genome as infectious bacterial artificial chromosomes for analysis of viral gene functions. J. Virol. Methods 2009, 158, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Baigent, S.J.; Petherbridge, L.J.; Smith, L.P.; Zhao, Y.; Chesters, P.M.; Nair, V.K. Herpesvirus of turkey reconstituted from bacterial artificial chromosome clones induces protection against Marek’s disease. J. Gen. Virol. 2006, 87, 769–776. [Google Scholar] [CrossRef]
- Sun, A.; Lawrence, P.; Zhao, Y.; Li, Y.; Nair, V.K.; Cui, Z. A BAC clone of MDV strain GX0101 with REV-LTR integration retained its pathogenicity. Chin. Sci. Bull. 2009, 54, 2641–2647. [Google Scholar] [CrossRef] [Green Version]
- Deruelle, M.J.; Favoreel, H.W. Keep it in the subfamily: The conserved alphaherpesvirus US3 protein kinase. J. Gen. Virol. 2011, 92, 18–30. [Google Scholar] [CrossRef]
- Lee, L.F.; Heidari, M.; Zhang, H.; Lupiani, B.; Reddy, S.M.; Fadly, A. Cell culture attenuation eliminates rMd5DeltaMeq-induced bursal and thymic atrophy and renders the mutant virus as an effective and safe vaccine against Marek’s disease. Vaccine 2012, 30, 5151–5158. [Google Scholar] [CrossRef]
- Dunn, J.R.; Silva, R.F. Ability of MEQ-deleted MDV vaccine candidates to adversely affect lymphoid organs and chicken weight gain. Avian Dis. 2012, 56, 494–500. [Google Scholar] [CrossRef]
- Liao, Y.; Reddy, S.M.; Khan, O.A.; Sun, A.; Lupiani, B. A Novel Effective and Safe Vaccine for Prevention of Marek’s Disease Caused by Infection with a Very Virulent Plus (vv+) Marek’s Disease Virus. Vaccines 2021, 9, 159. [Google Scholar] [CrossRef]
- Blondeau, C.; Chbab, N.; Beaumont, C.; Courvoisier, K.; Osterrieder, N.; Vautherot, J.F.; Denesvre, C. A full UL13 open reading frame in Marek’s disease virus (MDV) is dispensable for tumor formation and feather follicle tropism and cannot restore horizontal virus transmission of rRB-1B in vivo. Vet. Res. 2007, 38, 419–433. [Google Scholar] [CrossRef] [Green Version]
- Spatz, S.J.; Zhao, Y.; Petherbridge, L.; Smith, L.P.; Baigent, S.J.; Nair, V. Comparative sequence analysis of a highly oncogenic but horizontal spread-defective clone of Marek’s disease virus. Virus Genes 2007, 35, 753–766. [Google Scholar] [CrossRef]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.B.; Kim, D.Y.; Ko, J.H.; Kim, Y.S. Recent advances in the CRISPR genome editing tool set. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, S.; Teimoori, A.; Khanbabaei, H.; Tabasi, M. Harnessing CRISPR/Cas 9 System for manipulation of DNA virus genome. Rev. Med. Virol. 2019, 29, e2009. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef]
- Suenaga, T.; Kohyama, M.; Hirayasu, K.; Arase, H. Engineering large viral DNA genomes using the CRISPR-Cas9 system. Microbiol. Immunol. 2014, 58, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Quake, S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Sheng, J.; Trang, P.; Liu, F. Potential Application of the CRISPR/Cas9 System against Herpesvirus Infections. Viruses 2018, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.K.; Hu, W.; Khalili, K. The CRISPR/Cas9 genome editing methodology as a weapon against human viruses. Discov. Med. 2015, 19, 255–262. [Google Scholar]
- Van Diemen, F.R.; Lebbink, R.J. CRISPR/Cas9, a powerful tool to target human herpesviruses. Cell Microbiol. 2017, 19, e12694. [Google Scholar] [CrossRef]
- Wang, D.; Wang, X.W.; Peng, X.C.; Xiang, Y.; Song, S.B.; Wang, Y.Y.; Chen, L.; Xin, V.W.; Lyu, Y.N.; Ji, J.; et al. CRISPR/Cas9 genome editing technology significantly accelerated herpes simplex virus research. Cancer Gene Ther. 2018, 25, 93–105. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Pedrera, M.; Chang, P.; Baigent, S.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. A simple and rapid approach to develop recombinant avian herpesvirus vectored vaccines using CRISPR/Cas9 system. Vaccine 2018, 36, 716–722. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Sadigh, Y.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Generation of A Triple Insert Live Avian Herpesvirus Vectored Vaccine Using CRISPR/Cas9-Based Gene Editing. Vaccines 2020, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.; Ameen, F.; Sealy, J.E.; Sadeyen, J.R.; Bhat, S.; Li, Y.; Iqbal, M. Application of HDR-CRISPR/Cas9 and Erythrocyte Binding for Rapid Generation of Recombinant Turkey Herpesvirus-Vectored Avian Influenza Virus Vaccines. Vaccines 2019, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Vilela, J.; Rohaim, M.A.; Munir, M. Application of CRISPR/Cas9 in Understanding Avian Viruses and Developing Poultry Vaccines. Front. Cell Infect. Microbiol. 2020, 10, 581504. [Google Scholar] [CrossRef]
- Hagag, I.T.; Wight, D.J.; Bartsch, D.; Sid, H.; Jordan, I.; Bertzbach, L.D.; Schusser, B.; Kaufer, B.B. Abrogation of Marek’s disease virus replication using CRISPR/Cas9. Sci. Rep. 2020, 10, 10919. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Z.; Zhang, Y.; Yu, M.; Wang, S.; Gao, Y.; Liu, C.; Zhang, Y.; Gao, L.; Qi, X.; et al. Marek’s disease virus as a CRISPR/Cas9 delivery system to defend against avian leukosis virus infection in chickens. Vet. Microbiol. 2020, 242, 108589. [Google Scholar] [CrossRef] [PubMed]
- Challagulla, A.; Jenkins, K.A.; O’Neil, T.E.; Shi, S.; Morris, K.R.; Wise, T.G.; Paradkar, P.N.; Tizard, M.L.; Doran, T.J.; Schat, K.A. In Vivo Inhibition of Marek’s Disease Virus in Transgenic Chickens Expressing Cas9 and gRNA against ICP4. Microorganisms 2021, 9, 164. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature Sensitive (ts) Mutant | Marker Assisted Site-Directed Mutagenesis | Overlapping Cosmid Clones | BAC Clone (RecA or Red-Based Homologous Recombination) | CRISPR/CAS9 System | |
|---|---|---|---|---|---|
| Pros | 1. Allows to generate large number of mutants at once | 1. Allows site specific manipulation | 1. Allows site specific manipulation 2. No selection and purification steps are needed | 1. Capable of harboring large DNA fragments 2. Easy to manipulate using bacteria genetics 3. Can achieve both random and site-specific mutations 4. Easy to generate revertant BAC | 1. Easy to handle and efficient 2. Will not retain BAC sequences in the virus genome 3. Allows the simultaneous manipulation at different sites 4. Provides therapeutic potential for treatment |
| Cons | 1. Mutation frequency is low 2. Procedure is laborious 3. Difficult to precisely map the mutation site | 1. Low recombination efficacy 2. Need plaque for the purification 3. The inserted foreign gene may affect phenotype of the recombinant virus | 1. Difficult to handle large DNA fragments 2. Unwanted mutations may be introduced due to multiple recombination events 3. Difficult to construct revertant virus | 1. The large BAC DNA may shear during the manipulation process (mostly with RecA) 2. Unwanted recombination events or random mutations may occur (mostly with RecA) | 1. The need of PAM sequences may limit the target sites 2. The possibility of off-target may cause unwanted mutations 3. Needs multiple rounds of plaque purification |
| Method | MDV Strain | Manipulation | Main Findings | References |
|---|---|---|---|---|
| Marker assisted site-directed mutagenesis | RB-1B (vv) | Deletion of 4.5 kb sequences in US region of MDV genome | These genes are involved in virus replication, horizontal transmission, tumor formation, but not transformation | [36] |
| RB-1B (vv) | Deletion of vIL8 | vIL8 is important for MDV lytic infection but dispensable for transformation | [37] | |
| Overlapping cosmid clones | Md5 (vv) | Deletion of pp38 | pp38 is important MDV early cytolytic infection in lymphocytes but dispensable for virus growth in vitro, tumor formation in chickens and virus horizontal transmission | [38,39] |
| Md5 (vv) | Deletion of vIL8 | vIL8 is important for MDV early cytolytic infection in lymphoid organs, but dispensable for establishment of latency and virus horizontal transmission | [40,41] | |
| Md5 (vv) | Deletion of Meq | Meq is essential for tumor formation but dispensable for virus replication | [17] | |
| Md5 (vv) | Chimeric Meq mutants | Both homo- and heterodimerization of Meq are important for transformation of lymphocytes | [42,43] | |
| Md5 (vv) | Deletion of LORF11 | LORF11 is important for MDV replication and pathogenesis in chickens | [44] | |
| BAC clone | 584Ap80C (vv+, attenuated) | Deletion of 2 kb sequences in gB | gB is essential for cell-to-cell spread of MDV in vitro | [45] |
| RB-1B (vv) | Deletion of CtBP interaction domain in Meq | Meq-CtBP interaction is essential for MDV tumorigenesis | [46] | |
| RB-1B (vv) | Deletion of vTR | vTR is important for MDV induced T cell lymphoma | [16] | |
| RB-1B (vv) | Deletion of cluster 1 miRNAs and miR-M4 | Cluster 1 miRNAs, especially miR-M4, are important for MDV induced T cell lymphomas | [47] | |
| RB-1B (vv) | Deletion or mutation | UL13, UL44, UL47 and UL54 are essential for horizontal transmission of MDV | [48,49,50,51,52,53] | |
| 584Ap80C (vv+, attenuated), 686 (vv+) | Deletion of US3 and mutation of US3 kinase active site | US3 is involved in de-envelopment of perinuclear virion, actin stress fiber breakdown, antiapoptosis, MDV replication and gene expression | [54,55,56,57] | |
| 584Ap80C (vv+, attenuated) | Deletion of UL46 to UL49 | UL46, UL47 and UL48 genes are nonessential, but UL49 is essential, for growth of MDV | [58] | |
| RB-1B (vv), Md5 (vv) | Fusing of fluorescent protein to UL47, Meq and VP22 | Constructed fluorescent tagged viruses, which are valuable models to study MDV biology and pathogenesis | [59,60,61] | |
| CRISPR/Cas9 system | CVI988 (vaccine strain) | Deletion of Meq and pp38 | CRISPR/Cas9 system is applicable for MDV genome manipulation and gene function study | [62] |
| MDV transformed lymphoblastoid cell line | Deletion of pp38 and miRNAs | CRISPR/Cas9 system is applicable for MDV genome manipulation in MDV lymphoblastoid cell line | [63,64,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Y.; Bajwa, K.; Reddy, S.M.; Lupiani, B. Methods for the Manipulation of Herpesvirus Genome and the Application to Marek’s Disease Virus Research. Microorganisms 2021, 9, 1260. https://doi.org/10.3390/microorganisms9061260
Liao Y, Bajwa K, Reddy SM, Lupiani B. Methods for the Manipulation of Herpesvirus Genome and the Application to Marek’s Disease Virus Research. Microorganisms. 2021; 9(6):1260. https://doi.org/10.3390/microorganisms9061260
Chicago/Turabian StyleLiao, Yifei, Kanika Bajwa, Sanjay M. Reddy, and Blanca Lupiani. 2021. "Methods for the Manipulation of Herpesvirus Genome and the Application to Marek’s Disease Virus Research" Microorganisms 9, no. 6: 1260. https://doi.org/10.3390/microorganisms9061260