
Novel Siphoviridae Bacteriophages Infecting Bacteroides uniformis Contain Diversity Generating Retroelement


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Bacterial Strains from Human Fecal Sample
2.2. Phage Enrichment from Sterile Filtrate of Homogenized Fecal Sample
2.3. Phage Isolation from Enrichment Co-Cultures with Bacteroides
2.4. Preparation of Phage Stock Suspensions, EM Characterization and Host Range
2.5. Lysogen Formation Assay
2.6. Phage and Bacterial Genome Sequencing
2.7. Bacteriophage Genome Annotation
2.8. Phage Classification and Phylogenetic Analysis
2.9. Identification of Shared Homologous Proteins and Prophage Regions
2.10. Single Nucleotide Polymorphism (SNP) Analysis of Potential Phage Target Genes
2.11. Tandem Repeats Analysis with Direct Sequencing
2.12. Identification of the Isolated Bacteriophages in Metagenomes
2.13. Data Availability
3. Results
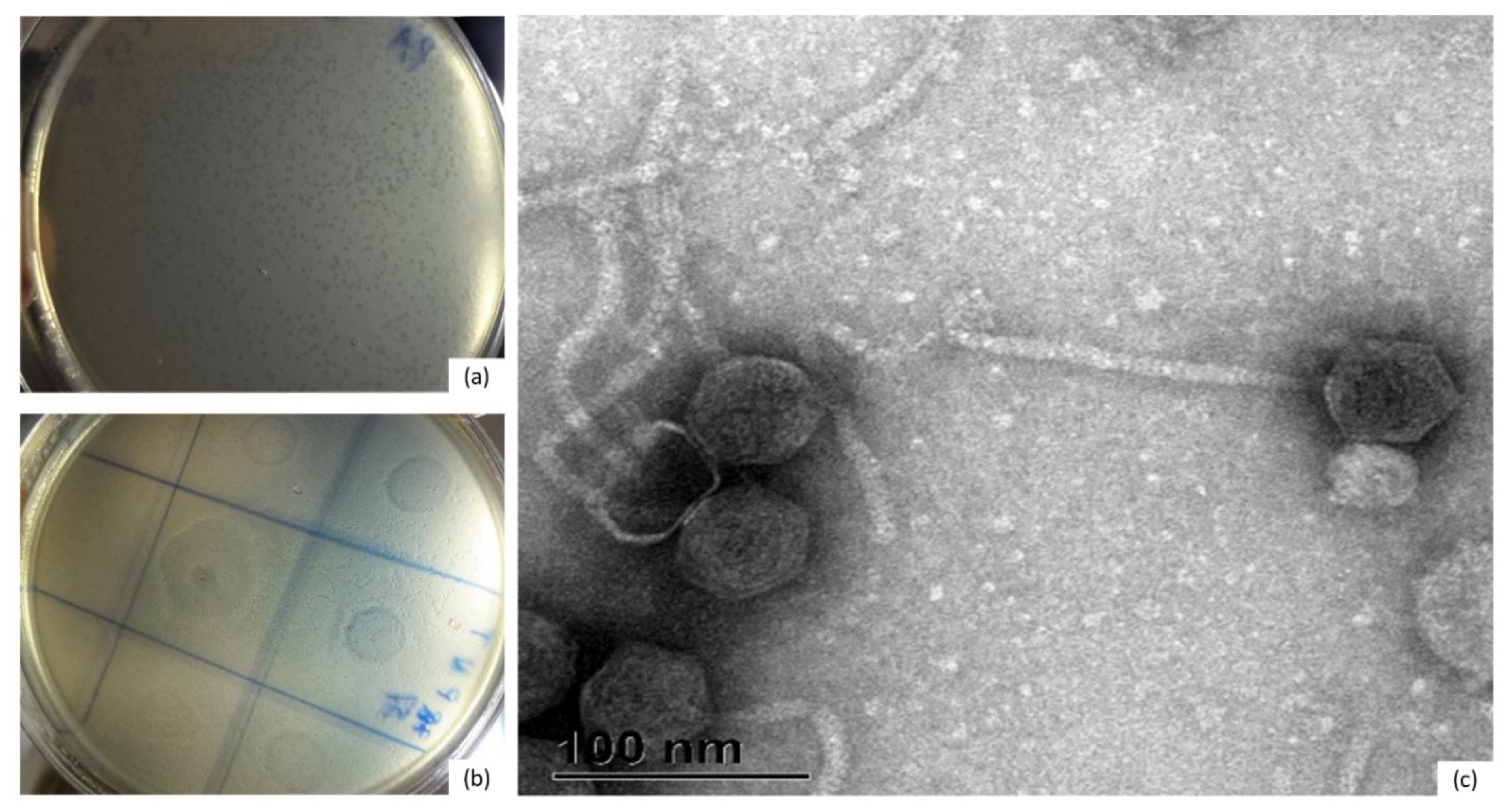
3.1. Isolation and Phenotypic Characterization of Phages Specific for Bacteroides uniformis
3.2. Novel B. uniformis Phages Show High Degree of Similarity to Each Other and Belong to a New Genus
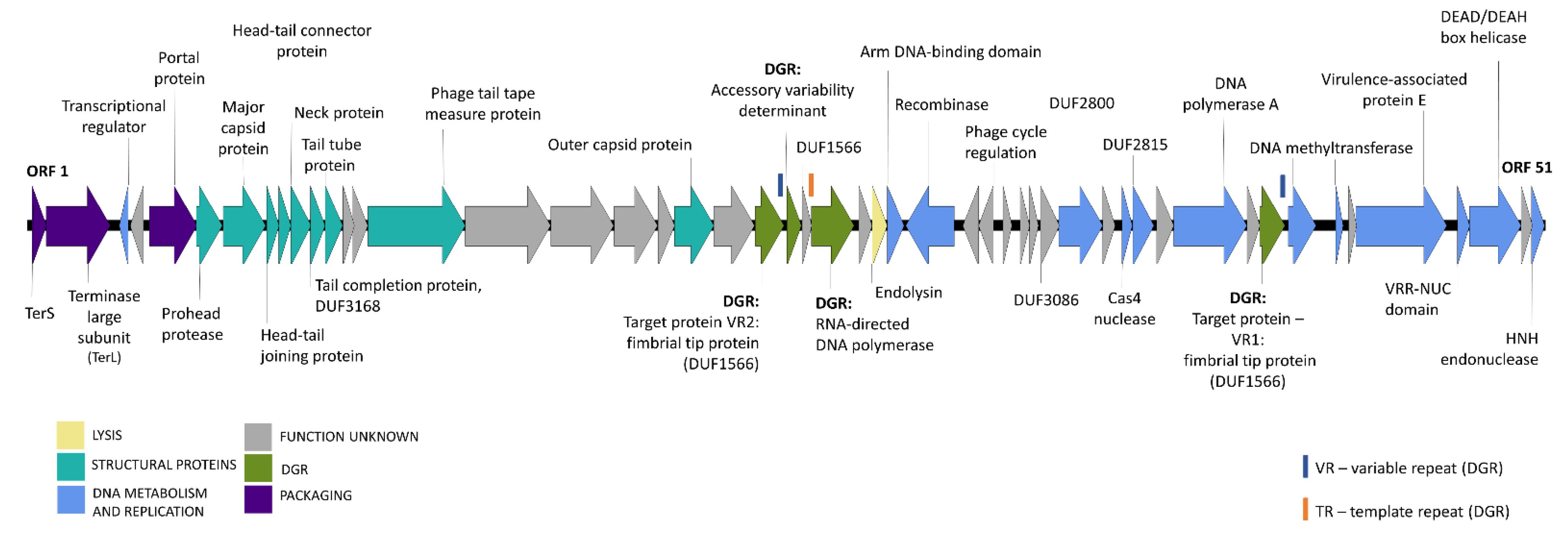
3.3. Genome Organization of Novel B. uniformis Phages
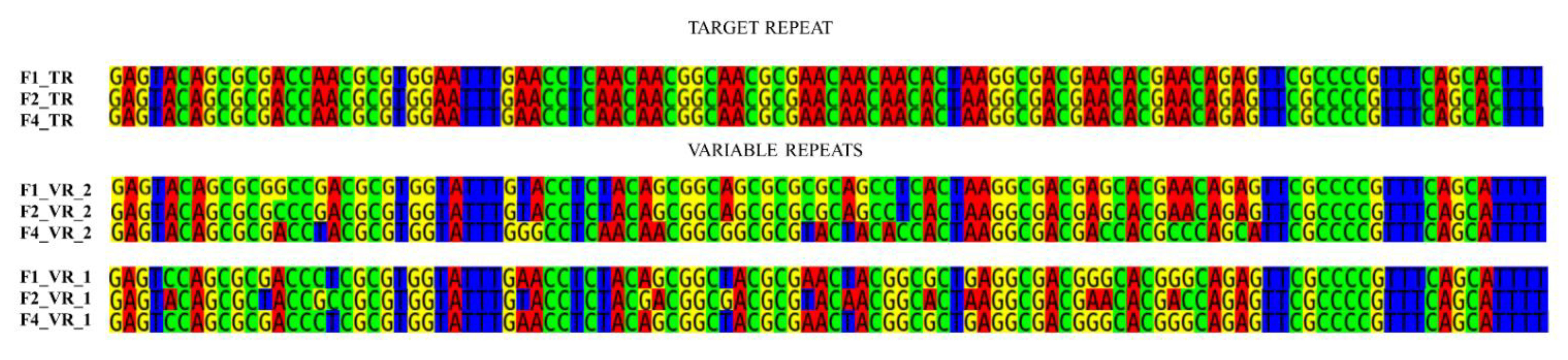
3.4. DGR Variability and Host Tropism
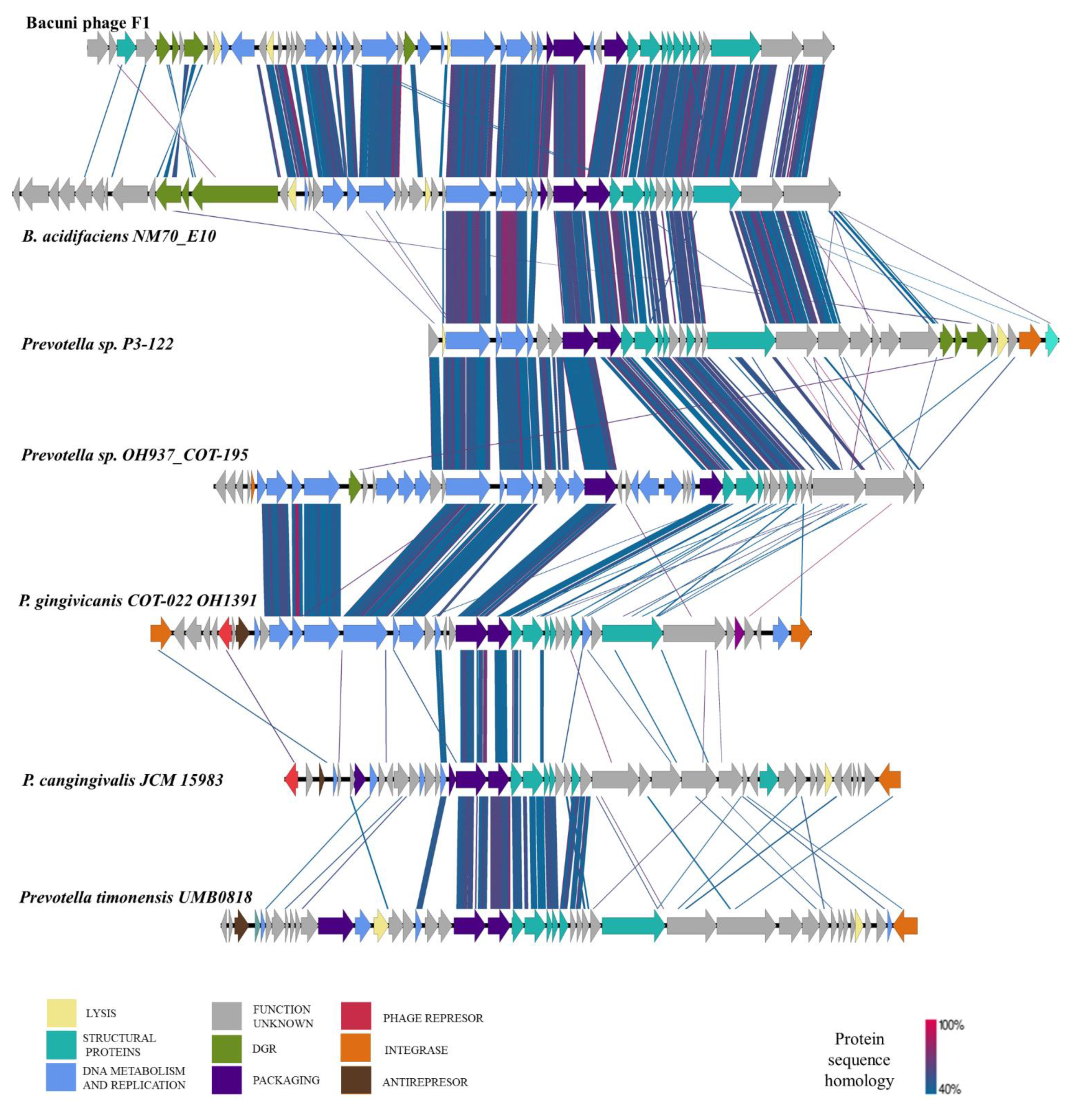
3.5. Bacuniphage Similarities to Other Phages and Prophages of Various Anaerobic Bacteria
3.6. Identification of Bacuni Phages in Human Gut Virome Database and in Associated Metagenomes
3.7. Changes of Host Susceptibility Pattern after Exposure to Bacuni Phage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sausset, R.; Petit, M.A.; Gaboriau-Routhiau, V.; de Paepe, M. New Insights into Intestinal Phages. Mucosal Immunol. 2020, 13, 205–215. [Google Scholar] [CrossRef] [Green Version]
- De Sordi, L.; Lourenço, M.; Debarbieux, L. “I Will Survive”: A Tale of Bacteriophage-Bacteria Coevolution in the Gut. Gut Microbe. 2019, 10, 92–99. [Google Scholar] [CrossRef]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral Communities of the Human Gut: Metagenomic Analysis of Composition and Dynamics. Mobile DNA 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The Human Gut Virome: Inter-Individual Variation and Dynamic Response to Diet The Human Gut Virome: Inter-Individual Variation and Dynamic Response to Diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Minot, S.; Bryson, A. Rapid Evolution of the Human Gut Virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive Expansion of Human Gut Bacteriophage Diversity. Cell 2021, 184, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A Highly Abundant Bacteriophage Discovered in the Unknown Sequences of Human Faecal Metagenomes. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of CrAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 Represents the Most Abundant Bacteriophage Family in the Human Gut and Infects Bacteroides Intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [Green Version]
- Hryckowian, A.J.; Merrill, B.D.; Porter, N.T.; van Treuren, W.; Nelson, E.J.; Garlena, R.A.; Russell, D.A.; Martens, E.C.; Sonnenburg, J.L. Bacteroides Thetaiotaomicron-Infecting Bacteriophage Isolates Inform Sequence-Based Host Range Predictions. Cell Host Microbe 2020, 28, 371–379. [Google Scholar] [CrossRef]
- Cornuault, J.K.; Petit, M.-A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; de Paepe, M. Phages Infecting Faecalibacterium Prausnitzii Belong to Novel Viral Genera That Help to Decipher Intestinal Viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef]
- Benler, S.; Cobián-Güemes, A.G.; McNair, K.; Hung, S.H.; Levi, K.; Edwards, R.; Rohwer, F. A Diversity-Generating Retroelement Encoded by a Globally Ubiquitous Bacteroides Phage 06 Biological Sciences 0605 Microbiology. Microbiome 2018, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages Infect Prevotella and Variants Are Widespread in Gut Microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornuault, J.K.; Moncaut, E.; Loux, V.; Mathieu, A.; Sokol, H.; Petit, M.-A.; de Paepe, M. The Enemy from within: A Prophage of Roseburia Intestinalis Systematically Turns Lytic in the Mouse Gut, Driving Bacterial Adaptation by CRISPR Spacer Acquisition. ISME J. 2020, 14, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, S.A.; Layton, A.C.; Ripp, S.; Williams, D.; Sayler, G.S. Genome Sequence of the Bacteroides Fragilis Phage ATCC 51477-B1. Virol. J. 2008, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, L.A.; Caplin, J.; Dedi, C.; Diston, D.; Cheek, E.; Bowler, L.; Taylor, H.; Ebdon, J.; Jones, B.V. Comparative (Meta)Genomic Analysis and Ecological Profiling of Human Gut-Specific Bacteriophage ΦB124-14. PLoS ONE 2012, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.A.; Kelly, W.J.; Altermann, E.; Leahy, S.C.; Minchin, C.; Ouwerkerk, D.; Klieve, A.V. Toward Understanding Phage: Host Interactions in the Rumen; Complete Genome Sequences of Lytic Phages Infecting Rumen Bacteria. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pacífico, C.; Hilbert, M.; Sofka, D.; Dinhopl, N.; Pap, I.J.; Aspöck, C.; Carriço, J.A.; Hilbert, F. Natural Occurrence of Escherichia Coli-Infecting Bacteriophages in Clinical Samples. Front. Microbiol. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Origins and Challenges of Viral Dark Matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Bianciotto, V.; Bondi, C.; Minerdi, D.; Sironi, M.; Tichy, H.V.; Bonfante, P. An Obligately Endosymbiotic Mycorrhizal Fungus Itself Harbors Obligately Intracellular Bacteria. Chemtracts 1998, 11, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput RRNA Analysis. Nucleic Acids Res. 2014, 42, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingett, S.W.; Andrews, S. Fastq Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000 Res. 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinform. Oxf. Engl. 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated Classification of Tailed Bacteriophages According to Their Neck Organization. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, F.; Ye, Y. MyDGR: A Server for Identification and Characterization of Diversity-Generating Retroelements. Nucleic Acids Res. 2019, 47, 289–294. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a Computational Approach to Classifying the Lifestyle of Phages. Bioinform. Oxf. Engl. 2012, 28, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.; Roux, S.; Sullivan, M.B. VConTACT: An IVirus Tool to Classify Double-Stranded DNA Viruses That Infect Archaea and Bacteria. PeerJ 2017, 5, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinform. Oxf. Engl. 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, Annotating and Comparing Sequences Stored in a Relational Database. Bioinform. Oxf. Engl. 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinform. Oxf. Engl. 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate Paired Shotgun Read Merging via Overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinform. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile Genetic Elements, Update 2010. Nucleic Acids Res. 2009, 38, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Belcaid, M.; Bergeron, A.; Poisson, G. The Evolution of the Tape Measure Protein: Units, Duplications and Losses. BMC Bioinform. 2011, 12, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, Z.; Lünse, C.E.; Corbino, K.A.; Ames, T.D.; Nelson, J.W.; Roth, A.; Perkins, K.R.; Sherlock, M.E.; Breaker, R.R. Detection of 224 Candidate Structured RNAs by Comparative Analysis of Specific Subsets of Intergenic Regions. Nucleic Acids Res. 2017, 45, 10811–10823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Gingery, M.; Abebe, M.; Arambula, D.; Czornyj, E.; Handa, S.; Khan, H.; Liu, M.; Pohlschroder, M.; Shaw, K.L.; et al. Diversity-Generating Retroelements: Natural Variation, Classification and Evolution Inferred from a Large-Scale Genomic Survey. Nucleic Acids Res. 2018, 46, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arambula, D.; Wong, W.; Medhekar, B.A.; Guo, H.; Gingery, M.; Czornyj, E.; Liu, M.; Dey, S.; Ghosh, P.; Miller, J.F. Surface Display of a Massively Variable Lipoprotein by a Legionella Diversity-Generating Retroelement. Proc. Natl. Acad. Sci. USA 2013, 110, 8212–8217. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Shoji, M.; Shibata, S.; Naito, M.; Sato, K.; Elsliger, M.-A.; Grant, J.C.; Axelrod, H.L.; Chiu, H.-J.; Farr, C.L.; et al. A Distinct Type of Pilus from the Human Microbiome. Cell 2016, 165, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y. Identification of Diversity-Generating Retroelements in Human Microbiomes. Int. J. Mol. Sci. 2014, 15, 14234–14246. [Google Scholar] [CrossRef]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.C.; Zablocki, O.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Human Gut Virome Database. bioRxiv 2019, 655910. [Google Scholar] [CrossRef]
- Rampelli, S.; Turroni, S.; Schnorr, S.L.; Soverini, M.; Quercia, S.; Barone, M.; Castagnetti, A.; Biagi, E.; Gallinella, G.; Brigidi, P.; et al. Characterization of the Human DNA Gut Virome across Populations with Different Subsistence Strategies and Geographical Origin. Environ. Microbiol. 2017, 19, 4728–4735. [Google Scholar] [CrossRef]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; van Ranst, M.; Matthijnssens, J. Gut Virome Analysis of Cameroonians Reveals High Diversity of Enteric Viruses, Including Potential Interspecies Transmitted Viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Batto, J.; Bertalan, M.; Borruel, N.; Casellas, F.; Costea, P.I.; Hildebrand, F.; et al. Enterotypes in the Landscape of Gut Microbial Community Composition. Nature 2013, 3, 1–12. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahnic, A.; Rupnik, M. Different Host Factors Are Associated with Patterns in Bacterial and Fungal Gut Microbiota in Slovenian Healthy Cohort. PLoS ONE 2018, 13, e0209209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Deora, R.; Doulatov, S.R.; Gingery, M.; Eiserling, F.A.; Preston, A.; Maskell, D.J.; Simons, R.W.; Cotter, P.A.; Parkhill, J.; et al. Reverse Transcriptase-Mediated Tropism Switching in Bordetella Bacteriophage. Science 2002, 295, 2091–2094. [Google Scholar] [CrossRef]
- Doulatov, S.; Hodes, A.; Dal, L.; Mandhana, N.; Liu, M.; Deora, R.; Simons, R.W.; Zimmerly, S.; Miller, J.F. Tropism Switching in Bordetella Bacteriophage Defines a Family of Diversity-Generating Retroelements. Nature 2004, 431, 476–481. [Google Scholar] [CrossRef]
- Cieplak, T.; Soffer, N.; Sulakvelidze, A.; Sandris, D. A Bacteriophage Cocktail Targeting Escherichia Coli Reduces E. Coli in Simulated Gut Conditions, While Preserving a Non-Targeted Representative Commensal Normal Microbiota. Gut Microbe. 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dalmasso, M.; Strain, R.; Neve, H.; Franz, C.M.A.P.; Cousin, F.J.; Ross, R.P.; Hill, C. Three New Escherichia Coli Phages from the Human Gut Show Promising Potential for Phage Therapy. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krylov, V.; Shaburova, O.; Krylov, S.; Pleteneva, E. A Genetic Approach to the Development of New Therapeutic Phages to Fight Pseudomonas Aeruginosa in Wound Infections. Viruses 2012, 5, 15–53. [Google Scholar] [CrossRef]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Paul Ross, R. Movers and Shakers: Influence of Bacteriophages in Shaping the Mammalian Gut Microbiota. Gut Microbe. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Tetz, G.V.; Ruggles, K.V.; Zhou, H.; Heguy, A.; Tsirigos, A.; Tetz, V. Bacteriophages as Potential New Mammalian Pathogens. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | F1 | F2 | F3 and F4 |
|---|---|---|---|
| Bacterial host | Bacteroides uniformis MB18-33 | Bacteroides uniformis MB18-33 | Bacteroides uniformis MB18-80 |
| No. of predicted ORFs | 51 | 51 | 50 |
| Assembled genome length (bp) | 40,421 | 40,653 | 40,112 |
| G + C content (%) | 51.8 | 51.7 | 51.7 |
| Genetic differences (compared to F1) | Reference | 16 SNPs in the DGR, of these 15 in the secondary VR and 57 bp insertion in RT gene | 24 SNPs, 18 in DGR primary VR RT identical to RT F1 |
| Host Strain | Source | Collection Date and Location | Region Length (bp) | No. of Bacuni Homologous Proteins/No. of ORFs | Coverage * (%) —nt Identity (%) | Genome Location and Biosample Accession |
|---|---|---|---|---|---|---|
| Bacteroides acidifaciens NM70_E10 | Mus musculus, colon and cecum | 2016, Toronto, Canada | 44,986 | 28/48 | 45% —71.06% | Node 8 (64,227,109,212) SAMN10878312 |
| Prevotella sp. P3-122 | Sus scrofa domesticus, feces | 2014, Slovenia: pig farm Ihan | 34,280 | Contig 46: 21/35, Contig 76: 5/15 | 46% —72.82% | Contig 46 (4,434,078,619) Contig 76 (112,007) SAMN07431220 |
| Prevotella sp. OH937_COT-195 | Canis lupus, dog mouth | 2012, Leicestershire, UK | 38,640 | 17/47 | 28% —71.82% | Scaffold20 (316,337,390) SAMN10478691 |
| Porphyromonas gingivicanis COT-022 OH1391 | Canis lupus, dog mouth | 2012, Leicestershire, UK | 35,922 | 11/39 | 23% —70.63% | Contig 6 (1,637,952,300) SAMN03004338 |
| Porphyromonas cangingivalis JCM 15983 | n.a. | 2014, The University of Tokyo | 33,481 | 17/46 | 33% —67.78% | Node 1 (310,636,586) SAMD00003336 |
| Prevotella timonensis UMB0818 | Homo sapiens, catheter | 2015, USA: Maywood, IL | 37,867 | 16/47 | 4% —69.19% | Node 1 (5,617,794,272) SAMN07511428 |
| Putative Functionof B. uniformis MB18-80 Protein | NCBI Accession * of Closest BLASTp Hit | SNP in B. uniformis MB18-80 K (Immune) | SNP in B. uniformis MB18-80 PH (Switched Tropism) |
|---|---|---|---|
| Type I restriction-modification system specificity (S) subunit | WP_117795664.1, WP_118086673.1 | + | + |
| TonB-linked outer membrane protein, SusC receptor | EOS06643.1, WP_080597360.1 | + | − |
| Outer-membrane protein OmpA, DUF5082 | WP_034528676.1, WP_034528679.1 | − | + |
| Putative porin–exopolysaccharide biosynthesis protein YbjH | WP_034528957.1, WP_120141442.1, WP_147392574.1, WP_147392573.1 | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedžet, S.; Rupnik, M.; Accetto, T. Novel Siphoviridae Bacteriophages Infecting Bacteroides uniformis Contain Diversity Generating Retroelement. Microorganisms 2021, 9, 892. https://doi.org/10.3390/microorganisms9050892
Hedžet S, Rupnik M, Accetto T. Novel Siphoviridae Bacteriophages Infecting Bacteroides uniformis Contain Diversity Generating Retroelement. Microorganisms. 2021; 9(5):892. https://doi.org/10.3390/microorganisms9050892
Chicago/Turabian StyleHedžet, Stina, Maja Rupnik, and Tomaž Accetto. 2021. "Novel Siphoviridae Bacteriophages Infecting Bacteroides uniformis Contain Diversity Generating Retroelement" Microorganisms 9, no. 5: 892. https://doi.org/10.3390/microorganisms9050892