
Microbial Community Successional Changes in a Full-Scale Mesophilic Anaerobic Digester from the Start-Up to the Steady-State Conditions

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Full-Scale AD Plant
2.2. DNA Extraction
2.3. Real-Time PCR, qPCR
2.4. High-Throughput 16S rRNA Gene Sequencing
2.5. Bioinformatic Processing
2.6. Statistical Analysis
3. Results
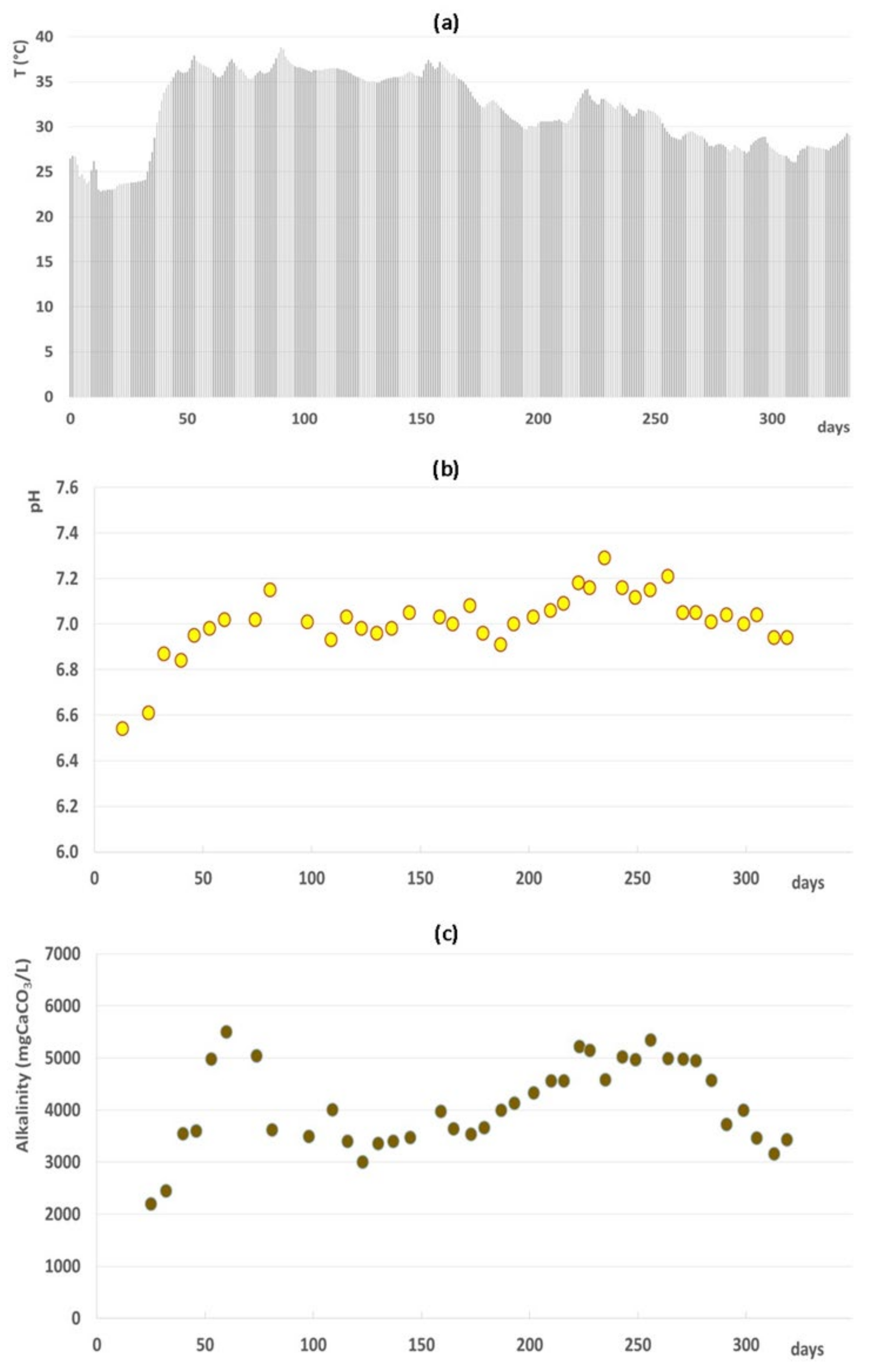
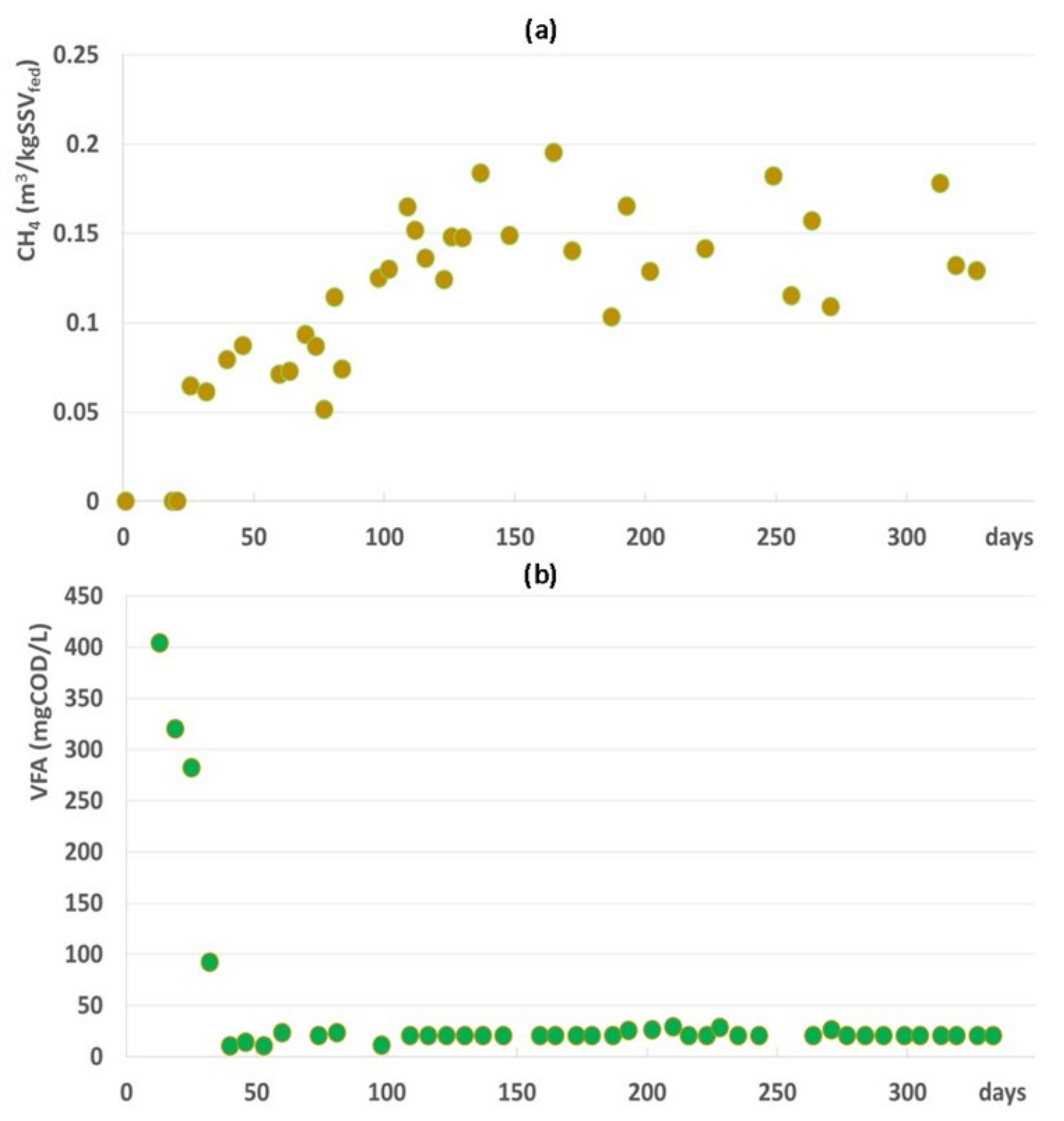
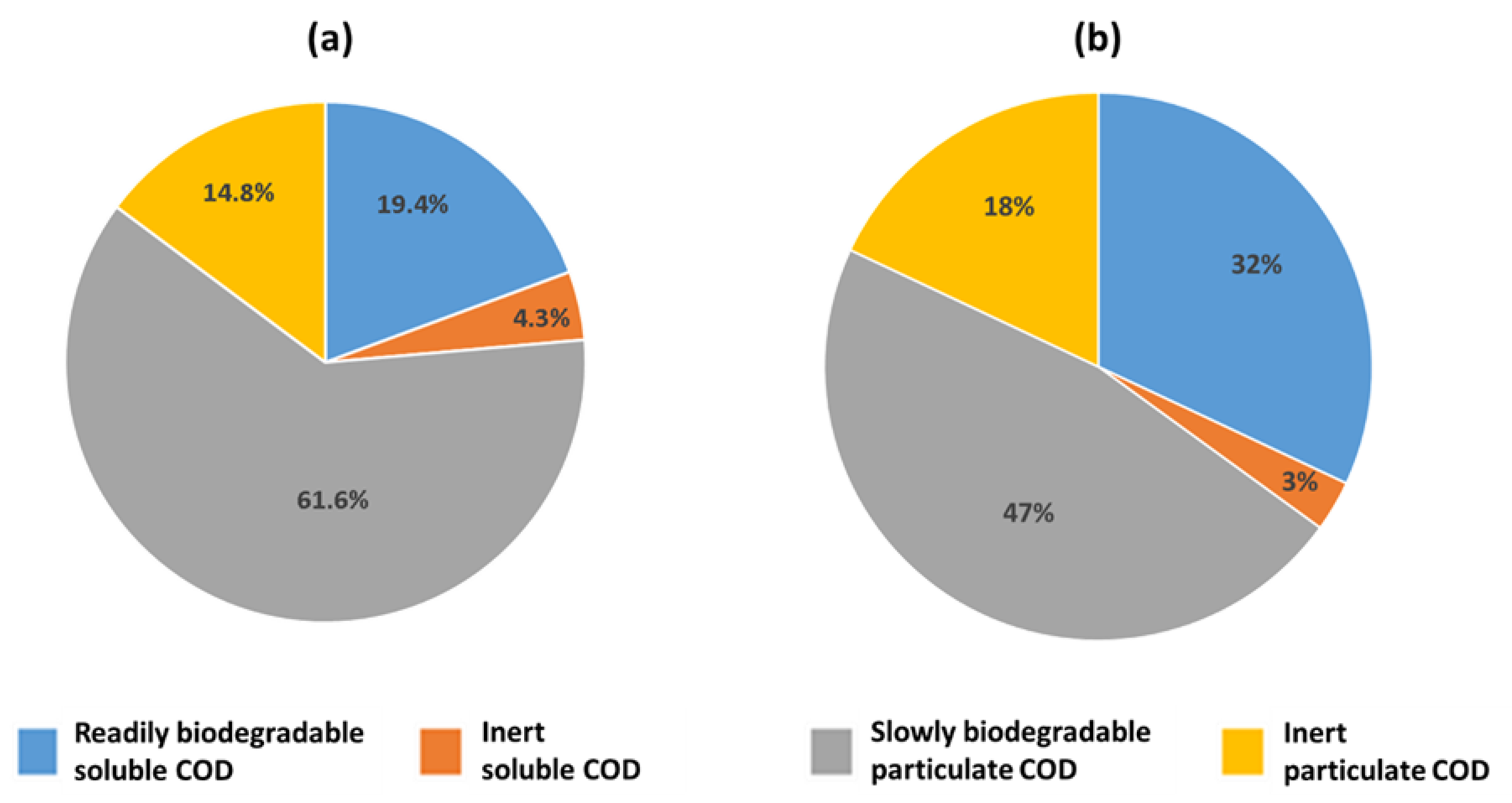
3.1. Full-Scale AD Plant
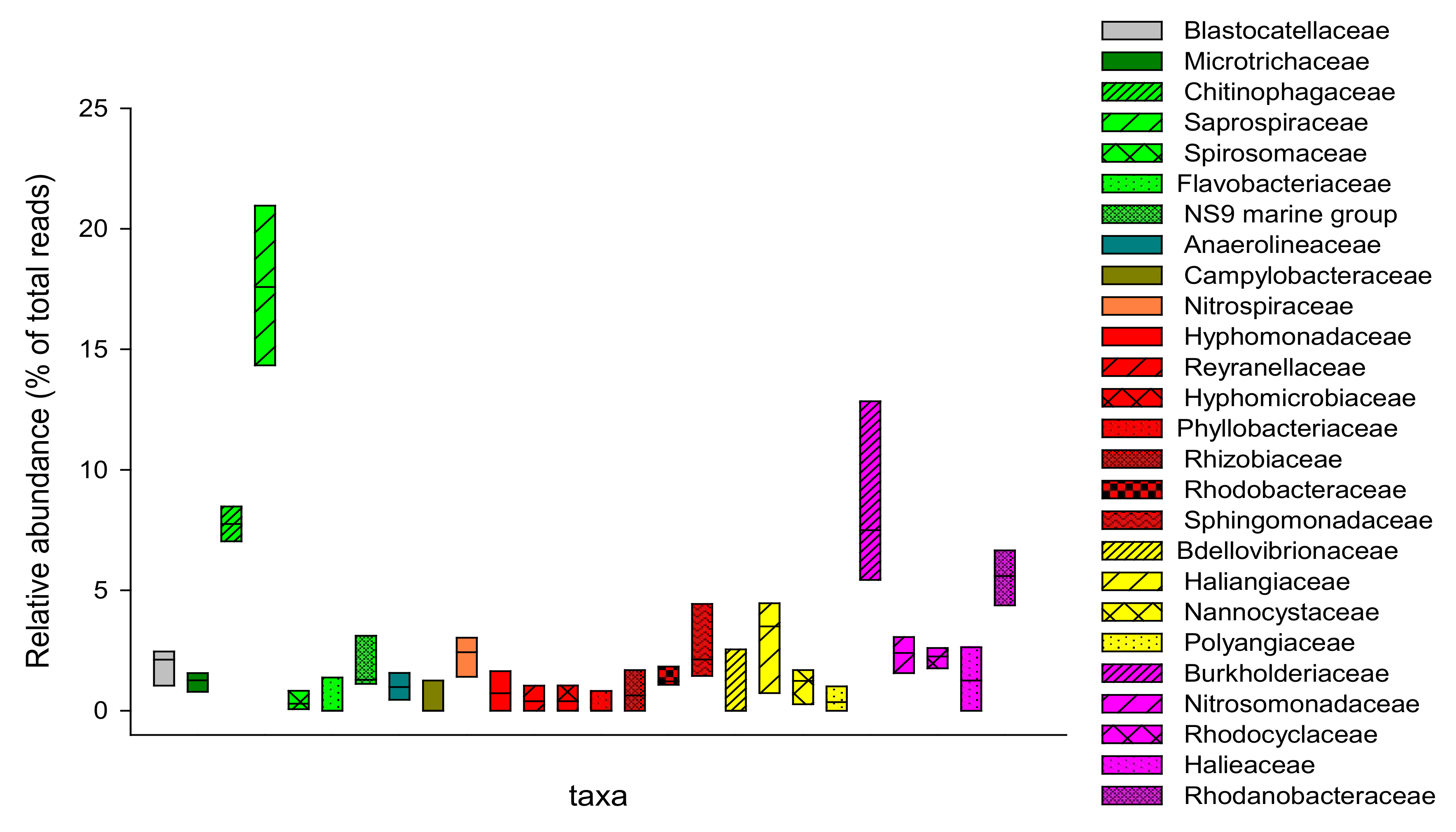
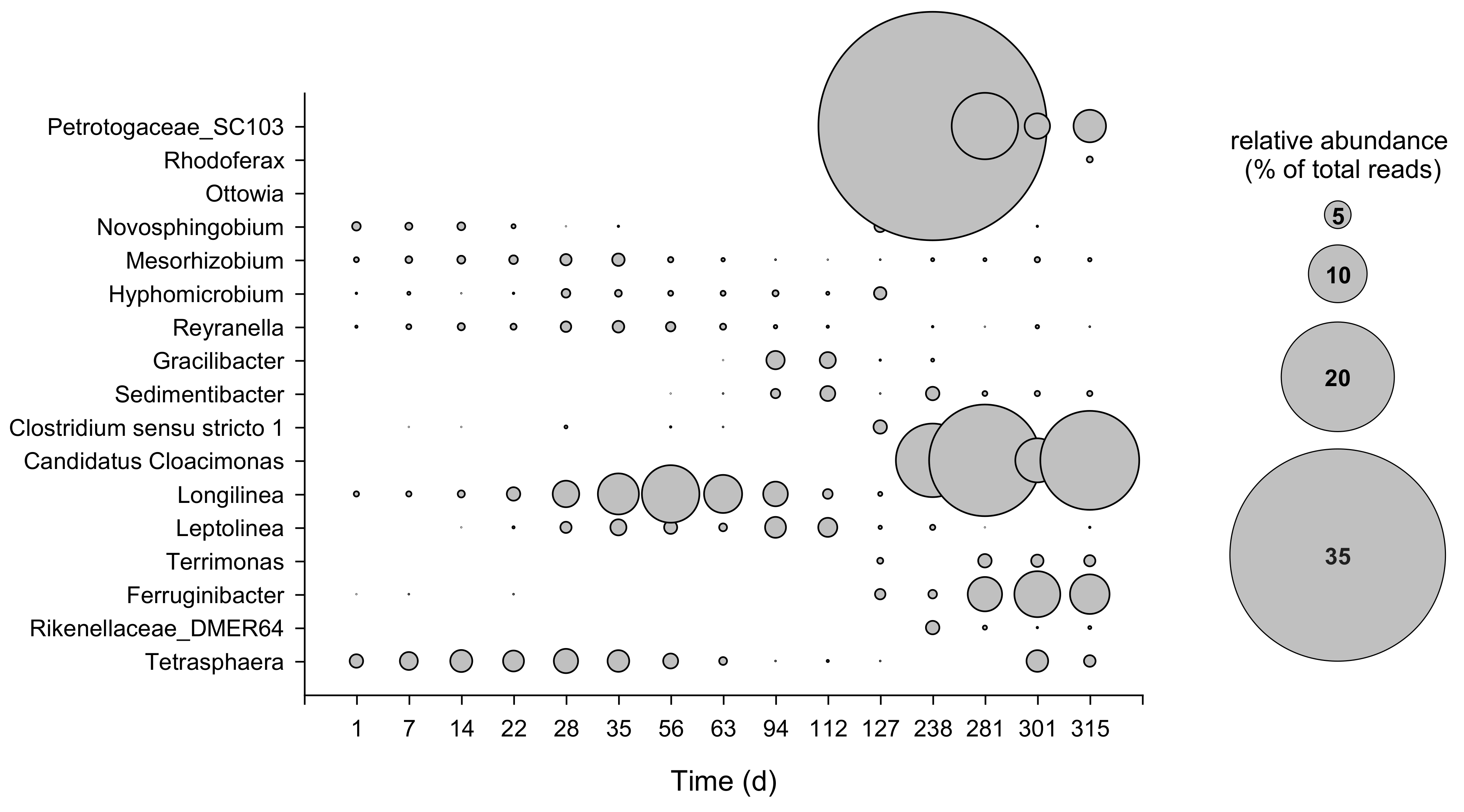
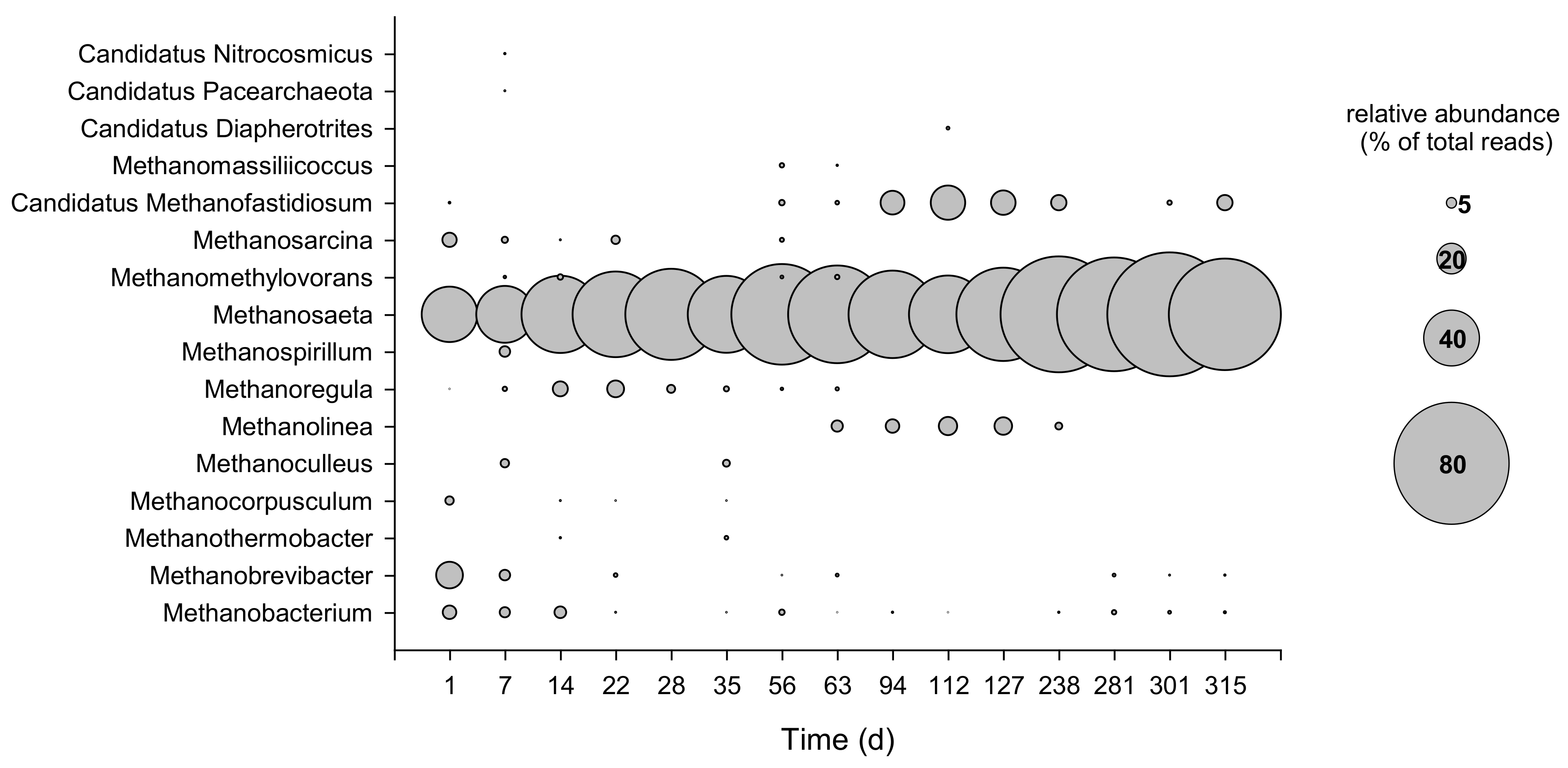
3.2. Microbial Composition of Secondary Sludge from Fusina WWTP
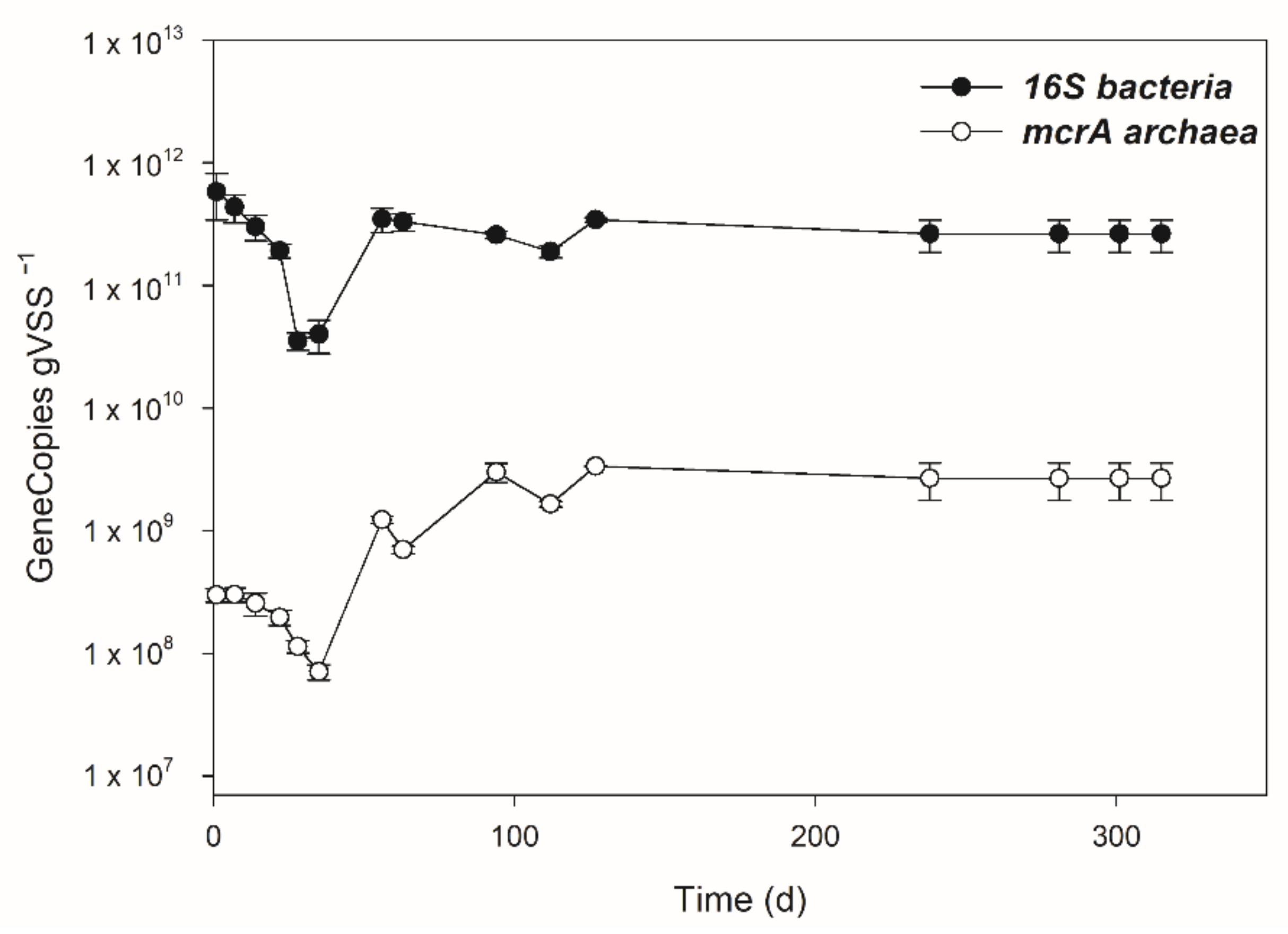
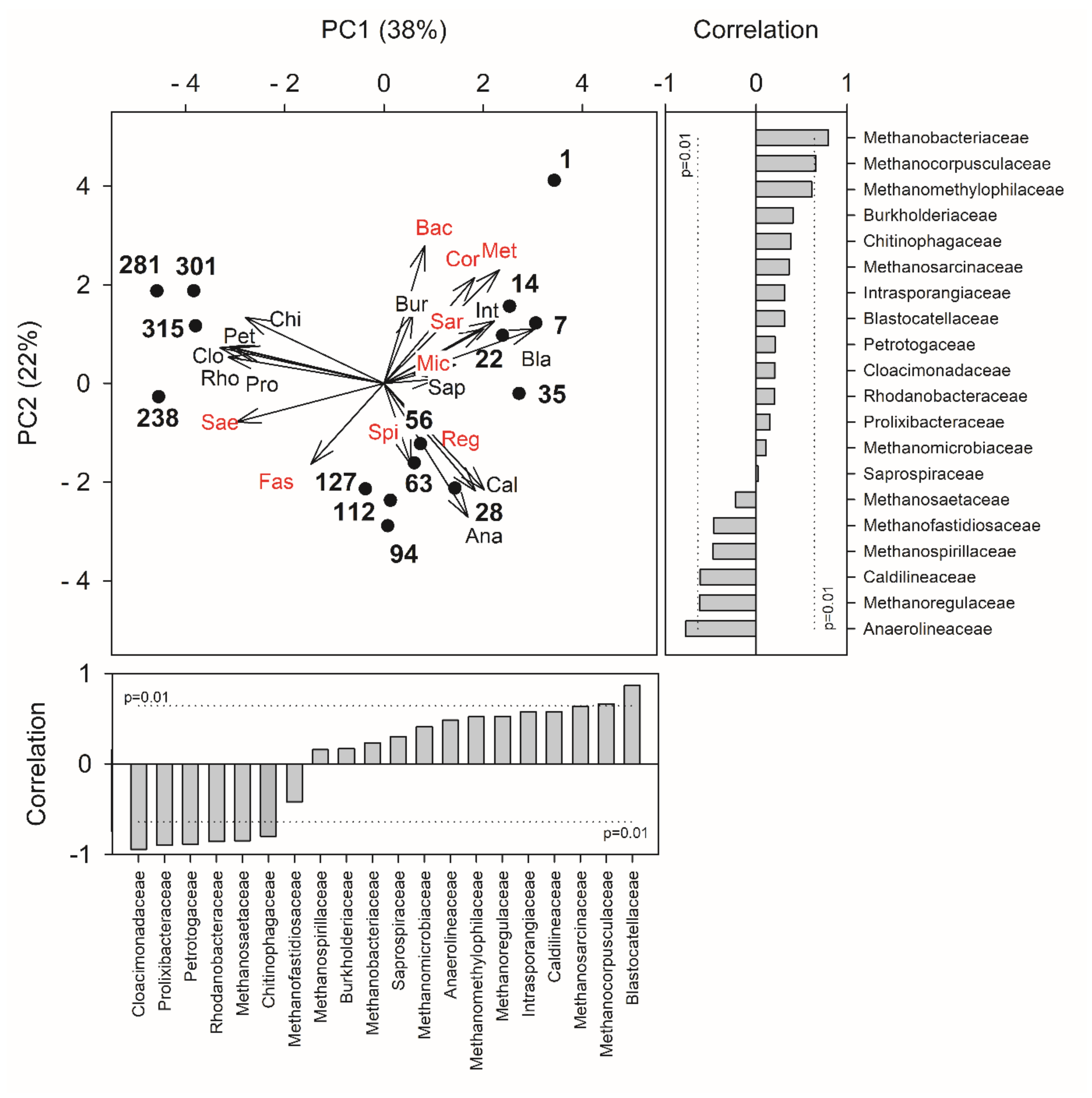
3.3. Dynamics of Bacterial and Archaeal Communities over the Operation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Vrieze, J.; Verstraete, W. Perspectives for Microbial Community Composition in Anaerobic Digestion: From Abundance and Activity to Connectivity. Environ. Microbiol. 2016, 18, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Appels, L.; Baeyens, J.; Degreve, J.; Dewil, R. Principles and Potential of the Anaerobic Digestion of Waste-Activated Sludge. Prog. Energy Combust. Sci. 2008, 34, 755–781. [Google Scholar] [CrossRef]
- Braguglia, C.M.; Gianico, A.; Mininni, G. ROUTES: Innovative Solutions for Municipal Sludge Treatment and Management. Rev. Environ. Sci. Biotechnol. 2011, 11, 11–17. [Google Scholar] [CrossRef]
- Weiland, P. Biogas Production: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2010, 85, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Vanwonterghem, I.; Jensen, P.D.; Dennis, P.G.; Hugenholtz, P.; Rabaey, K.; Tyson, G.W. Deterministic Processes Guide Long-Term Synchronised Population Dynamics in Replicate Anaerobic Digesters. ISME J. 2014, 8, 2015–2028. [Google Scholar] [CrossRef]
- Verstraete, W.; Wittebolle, L.; Heylen, K.; Vanparys, B.; De Vos, P.; Boon, N. Microbial Resource Management: The Road to Go for Environmental Biotechnology. Eng. Life Sci. 2007, 7, 117–126. [Google Scholar] [CrossRef]
- Wilkins, D.; Lu, X.; Shen, Z.; Chen, J.; Lee, P.K.H. Pyrosequencing of McrA and Archaeal 16S RRNA Genes Reveals Diversity and Substrate Preferences of Methanogen Communities in Anaerobic Digesters. Appl. Environ. Microbiol. 2015, 81, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, R.H.; McIlroy, S.J.; Kristensen, J.M.; Nierychlo, M.; Karst, S.M.; Dueholm, M.S.; Albertsen, M.; Nielsen, P.H. The Impact of Immigration on Microbial Community Composition in Full-Scale Anaerobic Digesters. Sci. Rep. 2017, 7, 9343. [Google Scholar] [CrossRef]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X. Global Diversity and Biogeography of Bacterial Communities in Wastewater Treatment Plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, C.; Al-soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 Pyrosequencing Analyses of Bacterial and Archaeal Richness in 21 Full-Scale Biogas Digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.J.; Knights, D.; Garcia, M.L.; Scalfone, N.B.; Smith, S.; Yarasheski, K.; Cummings, T.A.; Beers, A.R.; Knight, R.; Angenent, L.T. Bacterial Community Structures Are Unique and Resilient in Full-Scale Bioenergy Systems. Proc. Natl. Acad. Sci. USA 2011, 108, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Su, H.; Baeyens, J.; Tan, T. Reviewing the Anaerobic Digestion of Food Waste for Biogas Production. Renew. Sustain. Energy Rev. 2014, 38, 383–392. [Google Scholar] [CrossRef]
- Jablonski, S.J.; Łukaszewicz, M. Mathematical Modelling of Methanogenic Reactor Start-up: Importance of Volatile Fatty Acids Degrading Population. Bioresour. Technol. 2014, 174, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Carballa, M.; Regueiro, L.; Lema, J.M. ScienceDirect Microbial Management of Anaerobic Digestion: Exploiting the Microbiome-Functionality Nexus. Curr. Opin. Biotechnol. 2015, 33, 103–111. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yasuda, D.; Li, Y.; Kubota, K.; Harada, H.; Yu, H. Bioresource Technology Characterization of Start-up Performance and Archaeal Community Shifts during Anaerobic Self-Degradation of Waste-Activated Sludge. Bioresour. Technol. 2009, 100, 4981–4988. [Google Scholar] [CrossRef]
- Ike, M.; Inoue, D.; Miyano, T.; Tong, T.; Sei, K.; Soda, S.; Kadoshin, S. Microbial Population Dynamics during Startup of a Full-Scale Anaerobic Digester Treating Industrial Food Waste in Kyoto Eco-Energy Project. Bioresour. Technol. 2010, 101, 3952–3957. [Google Scholar] [CrossRef]
- Goux, X.; Calusinska, M.; Fossépré, M.; Benizri, E.; Delfosse, P. Start-up Phase of an Anaerobic Full-Scale Farm Reactor—Appearance of Mesophilic Anaerobic Conditions and Establishment of the Methanogenic Microbial Community. Bioresour. Technol. 2016, 212, 217–226. [Google Scholar] [CrossRef]
- Escudie, R.; Cresson, R.; Delgene, J.; Bernet, N. Control of Start-up and Operation of Anaerobic Biofilm Reactors: An Overview of 15 Years of Research. Water Res. 2010, 45, 1–10. [Google Scholar] [CrossRef]
- Ahring, B.K.; Sandberg, M.; Angelidaki, I. Volatile Fatty Acids as Indicators of Process Imbalance in Anaerobic Digestors. Appl. Microbiol. Biotechnol. 1995, 43, 559–565. [Google Scholar] [CrossRef]
- Stroot, P.G.; Mcmahon, K.D.; Mackie, R.I.; Raskin, L. Anaerobic Codigestion of Municipal Solid Waste and Biosolids under Various Mixing Conditions- I. Digester Performance. Water Res. 2001, 35, 1804–1816. [Google Scholar] [CrossRef]
- Kim, M.; Speece, R.E. Aerobic Waste Activated Sludge ( WAS ) for Start-up Seed of Mesophilic and Thermophilic Anaerobic Digestion Aerobic Waste Activated Sludge ( WAS ) for Start-up Seed of Mesophilic and Thermophilic Anaerobic Digestion. Water Res. 2002, 36, 3860–3866. [Google Scholar] [CrossRef]
- Lens, P.N.L.; De Poorter, M.P.; Cronenberg, C.C.; Verstraete, W.H. Sulfate Reducing and Methane Producing Bacteria In Aerobic Wastewater Treatment Systems. Water Res. 1995, 29, 871–880. [Google Scholar] [CrossRef]
- Ziganshina, E.E.; Bagmanova, A.R.; Khilyas, I.V.; Ziganshin, A.M. Assessment of a Biogas-Generating Microbial Community in a Pilot-Scale Anaerobic Reactor. J. Biosci. Bioeng. 2014, 117, 730–736. [Google Scholar] [CrossRef]
- Goberna, M.; Gadermaier, M.; Franke-Whittle, I.H.; Garcia, C.; Wett, B.; Insam, H. Start-up Strategies in Manure-Fed Biogas Reactors: Process Parameters and Methanogenic Communities. Biomass Bioenergy 2015, 75, 46–56. [Google Scholar] [CrossRef]
- Angenent, L.T.; Sung, S.; Raskin, L. Methanogenic Population Dynamics during Startup of a Full-Scale Anaerobic Sequencing Batch Reactor Treating Swine Waste. Water Res. 2002, 36, 4648–4654. [Google Scholar] [CrossRef]
- Tonanzi, B.; Gallipoli, A.; Gianico, A.; Montecchio, D.; Pagliaccia, P.; Di Carlo, M.; Rossetti, S.; Braguglia, C.M. Long-Term Anaerobic Digestion of Food Waste at Semi-Pilot Scale: Relationship between Microbial Community Structure and Process Performances. Biomass Bioenergy 2018, 118, 55–64. [Google Scholar] [CrossRef]
- Ritalahti, K.; Amos, B.K.; Sung, Y.; Wu, Q.; Koenigsberg, S.S.; Loffler, F.E. Quantitative PCR Targeting 16S RRNA and Reductive Dehalogenase Genes Simultaneously Monitors Multiple Dehalococcoides Strains. Appl. Environ. Microbiol. 2001, 72, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, L.M.; Regan, J.M. McrA-Targeted Real-Time Quantitative PCR Method to Examine Methanogen Communities. Appl. Environ. Microbiol. 2009, 75, 4435–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matturro, B.; Tandoi, V.; Rossetti, S. Different Activity Levels of Dehalococcoides Mccartyi Revealed by FISH and CARD-FISH under Non-Steady and Pseudo-Steady State Conditions. N. Biotechnol. 2013, 30, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Crognale, S.; Casentini, B.; Amalfitano, S.; Fazi, S.; Petruccioli, M.; Rossetti, S. Biological As(III) Oxidation in Biofilters by Using Native Groundwater Microorganisms. Sci. Total Environ. 2019, 651, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Gabriel, A.; Ghalith, A.; Alexander, H.; Alm, E.J.; Arumugam, M.; et al. QIIME 2: Reproducible, Interactive, Scalable, and Extensible Microbiome Data Science. PeerJ Prepr. 2018. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 5–7. [Google Scholar] [CrossRef]
- Riviere, D.; Desvignes, V.; Pelletier, E.; Rivie, D.; Guermazi, S.; Weissenbach, J.; Li, T.; Camacho, P.; Sghir, A. Towards the Definition of a Core of Microorganisms Involved in Anaerobic Digestion of Sludge. ISME J. 2009, 3, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Ma, H.; Hu, H.; Ren, H. Effect of Mixing Intensity on Hydrolysis and Acidification of Sewage Sludge in Two-Stage Anaerobic Digestion: Characteristics of Dissolved Organic Matter and the Key Microorganisms. Water Res. 2019, 148, 359–367. [Google Scholar] [CrossRef]
- Dohdoh, A.; Aboulfotoh, A. Start-up Performance of a Mesophilic Anaerobic Digester without External Inoculums. Environ. Prot. Eng. 2017, 43, 29–39. [Google Scholar] [CrossRef]
- Podmirseg, S.M.; Gadermaier, M.; Franke-Whittle, I.H.; Wett, B.; Insam, H.; Goberna, M. Prokaryotic Community Dynamics during the Start-Up of a Full-Scale Biogas Digester. J. Environ. Eng. 2016, 142, 1–11. [Google Scholar] [CrossRef]
- Li, A.; Chu, Y.; Wang, X.; Ren, L.; Yu, J.; Liu, X.; Yan, J.; Zhang, L.; Wu, S.; Li, S. A Pyrosequencing-Based Metagenomic Study of Methane-Producing Microbial Community in Solid-State Biogas Reactor. Biotechnol. Biofuels 2013, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Chen, Y.; Ndegwa, P. Association between Methane Yield and Microbiota Abundance in the Anaerobic Digestion Process: A Meta-Regression. Renew. Sustain. Energy Rev. 2021, 135, 110212. [Google Scholar] [CrossRef]
- Nobu, M.K.; Narihiro, T.; Kuroda, K.; Mei, R.; Liu, W. Chasing the Elusive Euryarchaeota Class WSA2: Genomes Reveal a Uniquely Fastidious Methyl- Reducing Methanogen. ISME J. 2016, 10, 2478–2487. [Google Scholar] [CrossRef]
- Tonanzi, B.; Braguglia, C.M.; Gallipoli, A.; Montecchio, D.; Pagliaccia, P.; Rossetti, S.; Gianico, A. Anaerobic Digestion of Mixed Urban Biowaste: The Microbial Community Shift towards Stability. N. Biotechnol. 2020, 55, 108–117. [Google Scholar] [CrossRef]
- Calusinska, M.; Goux, X.; Fossépré, M.; Muller, E.E.L.; Wilmes, P.; Delfosse, P. A Year of Monitoring 20 Mesophilic Full—Scale Bioreactors Reveals the Existence of Stable but Different Core Microbiomes in Bio - Waste and Wastewater Anaerobic Digestion Systems. Biotechnol. Biofuels 2018, 11, 196. [Google Scholar] [CrossRef]
- Briones, A.M.; Daugherty, B.J.; Angenent, L.T.; Rausch, K.D.; Tumbleson, M.E.; Raskin, L. Microbial Diversity and Dynamics in Multi- and Single-Compartment Anaerobic Bioreactors Processing Sulfate-Rich Waste Streams. Environ. Microbiol. 2007, 9, 93–106. [Google Scholar] [CrossRef]
- Wojcieszak, M.; Pyzik, A.; Poszytek, K.; Krawczyk, P.S.; Sobczak, A.; Lipinski, L.; Roubinek, O.; Palige, J.; Sklodowska, A.; Drewniak, L. Adaptation of Methanogenic Inocula to Anaerobic Digestion of Maize Silage. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Andreottola, G.; Foladori, P.; Ziglio, G. (Eds.) AA. VV. Dal Monitoraggio Convenzionale Al Controllo Avanzato Degli Impianti a Fanghi Attivi; SAN 4: Trento, Italy, 2005; ISBN 8884431174. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonanzi, B.; Crognale, S.; Gianico, A.; Della Sala, S.; Miana, P.; Zaccone, M.C.; Rossetti, S. Microbial Community Successional Changes in a Full-Scale Mesophilic Anaerobic Digester from the Start-Up to the Steady-State Conditions. Microorganisms 2021, 9, 2581. https://doi.org/10.3390/microorganisms9122581
Tonanzi B, Crognale S, Gianico A, Della Sala S, Miana P, Zaccone MC, Rossetti S. Microbial Community Successional Changes in a Full-Scale Mesophilic Anaerobic Digester from the Start-Up to the Steady-State Conditions. Microorganisms. 2021; 9(12):2581. https://doi.org/10.3390/microorganisms9122581
Chicago/Turabian StyleTonanzi, Barbara, Simona Crognale, Andrea Gianico, Stefano Della Sala, Paola Miana, Maria Chiara Zaccone, and Simona Rossetti. 2021. "Microbial Community Successional Changes in a Full-Scale Mesophilic Anaerobic Digester from the Start-Up to the Steady-State Conditions" Microorganisms 9, no. 12: 2581. https://doi.org/10.3390/microorganisms9122581