
Metagenomic Analysis of Anaerobic Microbial Communities Degrading Short-Chain Fatty Acids as Sole Carbon Sources

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Metagenome Sequencing
2.3. Bioinformatic Analysis
2.3.1. Basecalling, Quality Filtering, and Read Trimming
2.3.2. Analysis of Community Composition and Functional Potential
2.3.3. Reconstruction and Annotation of Metagenome-Assembled Genomes
3. Results
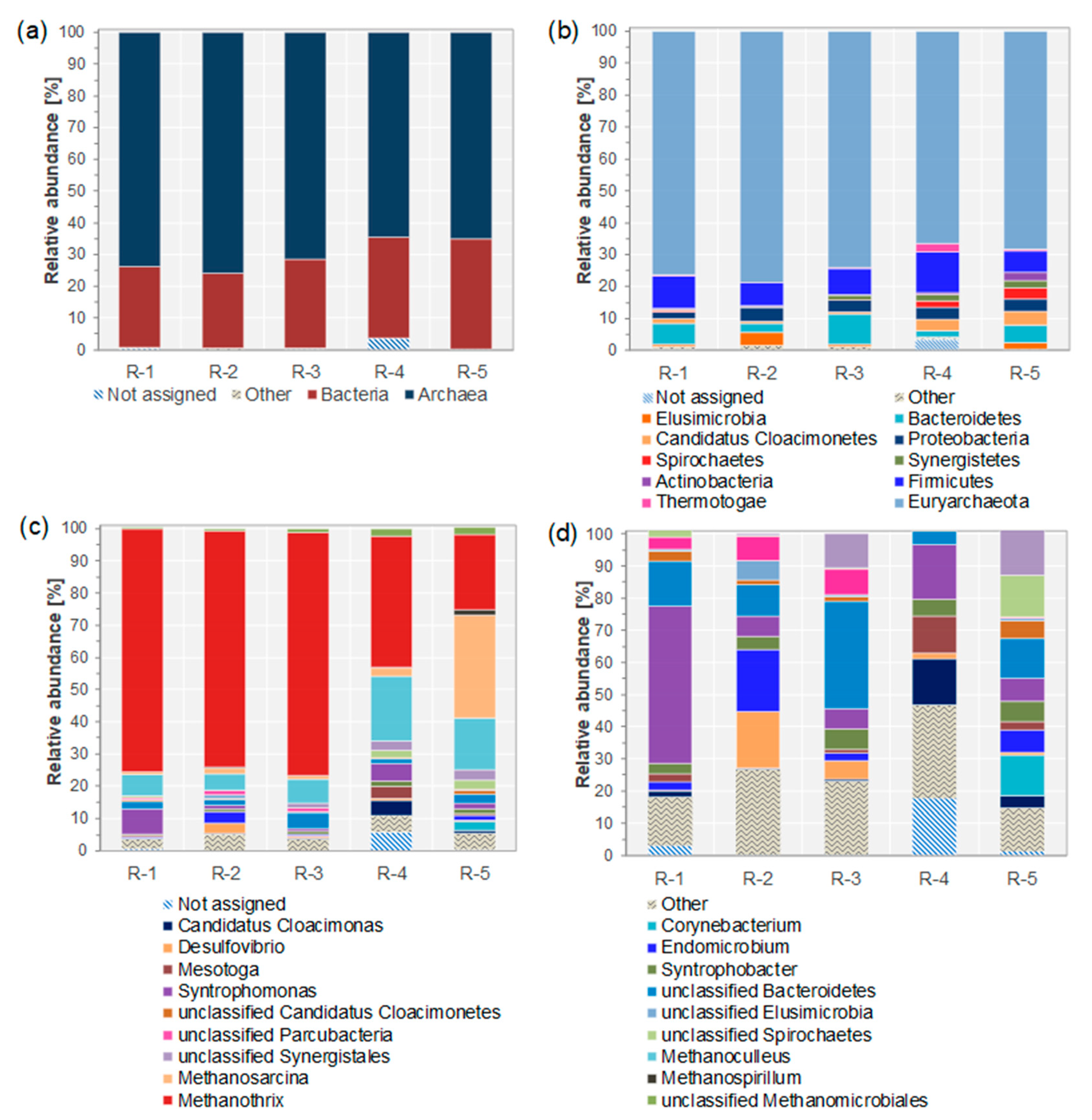
3.1. Impact of Feeding Regime and OLR on Microbial Community Composition
3.2. Combining Taxonomic and Functional Annotation
3.3. Metagenome-Assembled Genomes
3.3.1. MAGs Reconstructed from Illumina Reads Only
3.3.2. MAGs Based on the Hybrid Assembly Approach
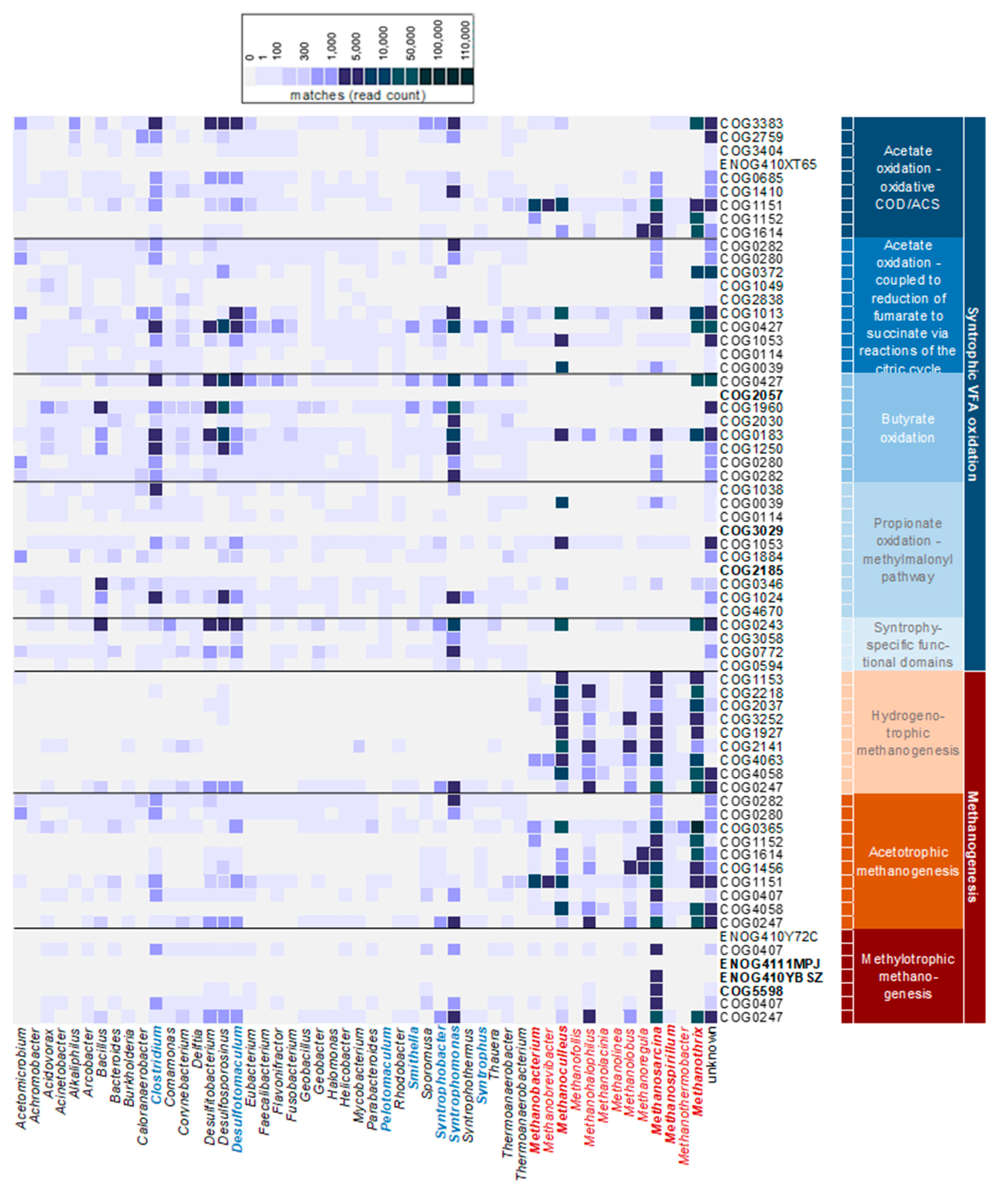
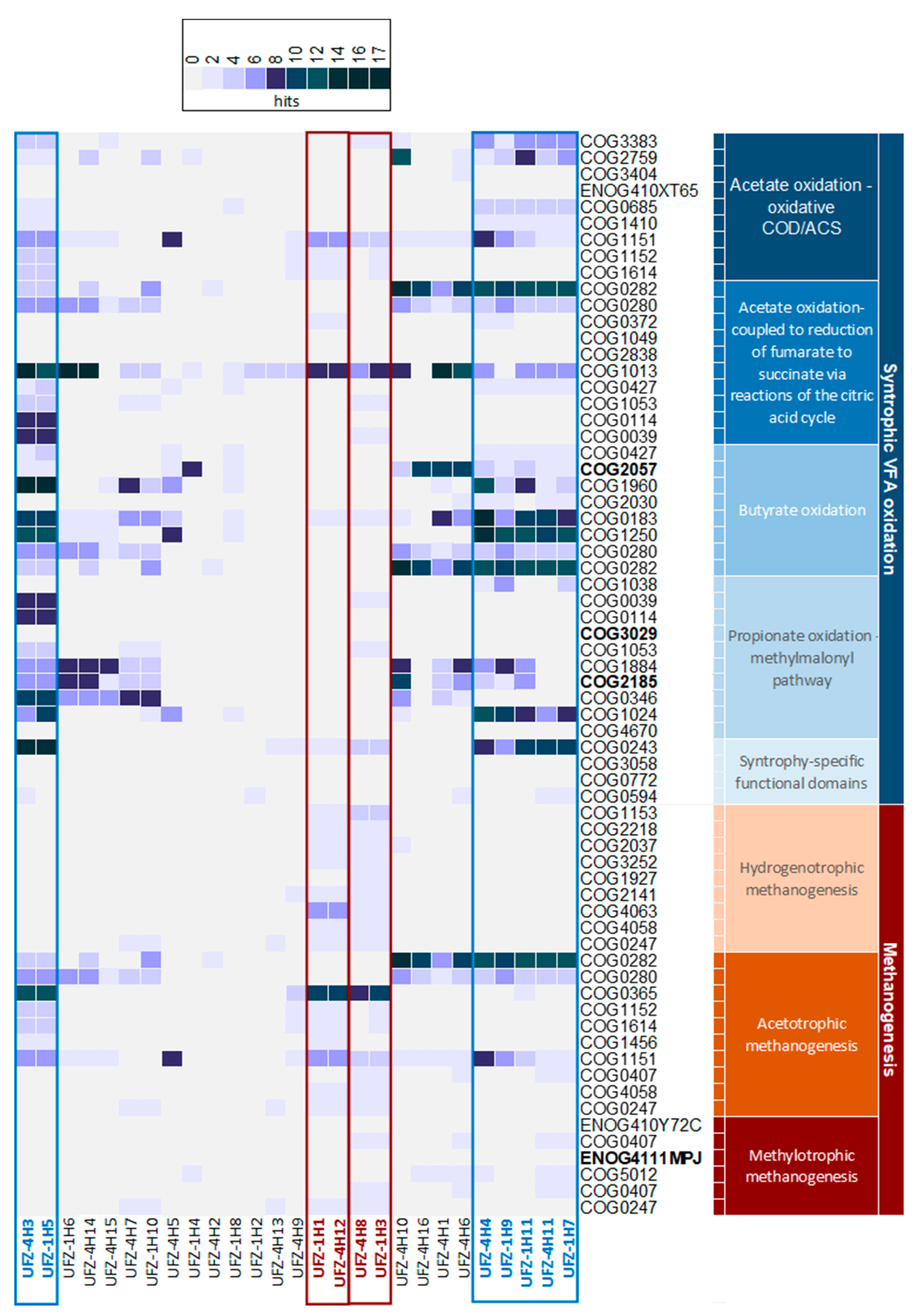
3.3.3. Pathways of Interest Encoded by MAGs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonk, F.; Popp, D.; Weinrich, S.; Sträuber, H.; Kleinsteuber, S.; Harms, H.; Centler, F. Intermittent Fasting for Microbes: How Discontinuous Feeding Increases Functional Stability in Anaerobic Digestion. Biotechnol. Biofuels 2018, 11, 274. [Google Scholar] [CrossRef] [PubMed]
- Vanwonterghem, I.; Jensen, P.D.; Dennis, P.G.; Hugenholtz, P.; Rabaey, K.; Tyson, G.W. Deterministic Processes Guide Long-Term Synchronised Population Dynamics in Replicate Anaerobic Digesters. ISME J. 2014, 8, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.W.; Song, K.-G.; Matiasek, M.G.; Wuertz, S. Microbial Community Dynamics in Replicate Membrane Bioreactors—Natural Reproducible Fluctuations. Water Res. 2009, 43, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Mulat, D.G.; Jacobi, H.F.; Feilberg, A.; Adamsen, A.P.S.; Richnow, H.-H.; Nikolausz, M. Changing Feeding Regimes to Demonstrate Flexible Biogas Production: Effects on Process Performance, Microbial Community Structure, and Methanogenesis Pathways. Appl. Env. Microb. 2016, 82, 438–449. [Google Scholar] [CrossRef]
- Vrieze, J.D.; Hennebel, T.; Boon, N.; Verstraete, W. Methanosarcina: The Rediscovered Methanogen for Heavy Duty Biomethanation. Bioresour. Technol. 2012, 112, 1–9. [Google Scholar] [CrossRef]
- Ziels, R.M.; Beck, D.A.C.; Stensel, H.D. Long-Chain Fatty Acid Feeding Frequency in Anaerobic Codigestion Impacts Syntrophic Community Structure and Biokinetics. Water Res. 2017, 117, 218–229. [Google Scholar] [CrossRef]
- Mauky, E.; Jacobi, H.F.; Liebetrau, J.; Nelles, M. Flexible Biogas Production for Demand-Driven Energy Supply—Feeding Strategies and Types of Substrates. Bioresour. Technol. 2015, 178, 262–269. [Google Scholar] [CrossRef]
- Vrieze, J.D.; Verstraete, W.; Boon, N. Repeated Pulse Feeding Induces Functional Stability in Anaerobic Digestion. Microb. Biotechnol. 2013, 6, 414–424. [Google Scholar] [CrossRef]
- Willeghems, G.; Buysse, J. Changing Old Habits: The Case of Feeding Patterns in Anaerobic Digesters. Renew. Energ. 2016, 92, 212–221. [Google Scholar] [CrossRef]
- Li, Y.; Ma, J.; Yuan, H.; Li, X. Effects of Feeding Regimes on Process Performance and Microbial Community Structure in Anaerobic Semi-Continuously Stirred Tank Reactors Treating Corn Stover. Waste Biomass Valorization 2022, 13, 1003–1014. [Google Scholar] [CrossRef]
- Gerardi, M.H. The Microbiology of Anaerobic Digesters. In Wastewater Microbiology Series; John Wiley & Sons Inc.: New Jersey, NJ, USA, 2003; pp. 121–125. [Google Scholar] [CrossRef]
- Hao, L.; Michaelsen, T.Y.; Singleton, C.M.; Dottorini, G.; Kirkegaard, R.H.; Albertsen, M.; Nielsen, P.H.; Dueholm, M.S. Novel Syntrophic Bacteria in Full-Scale Anaerobic Digesters Revealed by Genome-Centric Metatranscriptomics. ISME J. 2020, 14, 906–918. [Google Scholar] [CrossRef]
- Kirkegaard, R.H.; McIlroy, S.J.; Kristensen, J.M.; Nierychlo, M.; Karst, S.M.; Dueholm, M.S.; Albertsen, M.; Nielsen, P.H. The Impact of Immigration on Microbial Community Composition in Full-Scale Anaerobic Digesters. Sci. Rep. 2017, 7, 9343. [Google Scholar] [CrossRef]
- Ahring, B.K.; Sandberg, M.; Angelidaki, I. Volatile Fatty Acids as Indicators of Process Imbalance in Anaerobic Digestors. Appl. Microbiol. Biot. 1995, 43, 559–565. [Google Scholar] [CrossRef]
- Boe, K.; Batstone, D.J.; Steyer, J.-P.; Angelidaki, I. State Indicators for Monitoring the Anaerobic Digestion Process. Water Res. 2010, 44, 5973–5980. [Google Scholar] [CrossRef]
- Arumugam, K.; Bessarab, I.; Haryono, M.A.S.; Liu, X.; Zuniga–Montanez, R.E.; Roy, S.; Qiu, G.; Drautz–Moses, D.I.; Law, Y.Y.; Wuertz, S.; et al. Recovery of Complete Genomes and Non-Chromosomal Replicons from Activated Sludge Enrichment Microbial Communities with Long Read Metagenome Sequencing. Npj Biofilms Microbiomes 2021, 7, 23. [Google Scholar] [CrossRef]
- McInerney, M.J.; Struchtemeyer, C.G.; Sieber, J.; Mouttaki, H.; Stams, A.J.M.; Schink, B.; Rohlin, L.; Gunsalus, R.P. Physiology, Ecology, Phylogeny, and Genomics of Microorganisms Capable of Syntrophic Metabolism. Ann. NY Acad. Sci. 2008, 1125, 58–72. [Google Scholar] [CrossRef]
- Sousa, D.Z.; Smidt, H.; Alves, M.M.; Stams, A.J.M. Ecophysiology of Syntrophic Communities That Degrade Saturated and Unsaturated Long-Chain Fatty Acids: Ecophysiology of Syntrophic LCFA Degradation. FEMS Microbiol. Ecol. 2009, 68, 257–272. [Google Scholar] [CrossRef]
- Manzoor, S.; Schnürer, A.; Bongcam-Rudloff, E.; Müller, B. Genome-Guided Analysis of Clostridium ultunense and Comparative Genomics Reveal Different Strategies for Acetate Oxidation and Energy Conservation in Syntrophic Acetate-Oxidising Bacteria. Genes 2018, 9, 225. [Google Scholar] [CrossRef]
- Müller, B.; Sun, L.; Schnürer, A. First Insights into the Syntrophic Acetate-oxidizing Bacteria—A Genetic Study. MicrobiologyOpen 2013, 2, 35–53. [Google Scholar] [CrossRef]
- Westerholm, M.; Moestedt, J.; Schnürer, A. Biogas Production through Syntrophic Acetate Oxidation and Deliberate Operating Strategies for Improved Digester Performance. Appl. Energ. 2016, 179, 124–135. [Google Scholar] [CrossRef]
- Hattori, S. Syntrophic Acetate-Oxidizing Microbes in Methanogenic Environments. Microbes Environ. 2008, 23, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Plugge, C.M.; Henstra, A.M.; Worm, P.; Swarts, D.C.; Paulitsch-Fuchs, A.H.; Scholten, J.C.M.; Lykidis, A.; Lapidus, A.L.; Goltsman, E.; Kim, E.; et al. Complete Genome Sequence of Syntrophobacter fumaroxidans Strain (MPOB(T)). Stand. Genom. Sci. 2012, 7, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Felchner-Zwirello, M.; Winter, J.; Gallert, C. Interspecies Distances between Propionic Acid Degraders and Methanogens in Syntrophic Consortia for Optimal Hydrogen Transfer. Appl. Microbiol. Biot. 2013, 97, 9193–9205. [Google Scholar] [CrossRef] [PubMed]
- Plugge, C.M.; Dijkema, C.; Stams, A.J.M. Acetyl-CoA Cleavage Pathway in a Syntrophic Propionate Oxidizing Bacterium Growing on Fumarate in the Absence of Methanogens. FEMS Microbiol. Lett. 1993, 110, 71–76. [Google Scholar] [CrossRef]
- Bok, F.A.M.; de Stams, A.J.M.; Dijkema, C.; Boone, D.R. Pathway of Propionate Oxidation by a Syntrophic Culture of Smithella propionica and Methanospirillum hungatei. Appl. Environ. Microb. 2001, 67, 1800–1804. [Google Scholar] [CrossRef]
- Knoop, F. Der Abbau aromatischer Fettsäuren im Tierkörper. Beitr. Chem. Physiol. Pathol. 1904, 6, 150–162. [Google Scholar]
- Enzmann, F.; Mayer, F.; Rother, M.; Holtmann, D. Methanogens: Biochemical Background and Biotechnological Applications. AMB Express 2018, 8, 1. [Google Scholar] [CrossRef]
- Kurth, J.M.; Camp, H.J.M.O.; den Welte, C.U. Several Ways One Goal—Methanogenesis from Unconventional Substrates. Appl. Microbiol. Biot. 2020, 104, 6839–6854. [Google Scholar] [CrossRef]
- Crable, B.R.; Plugge, C.M.; McInerney, M.J.; Stams, A.J.M. Formate Formation and Formate Conversion in Biological Fuels Production. Enzym. Res. 2011, 2011, 532536. [Google Scholar] [CrossRef]
- Sikora, A.; Detman, A.; Mielecki, D.; Chojnacka, A.; Błaszczyk, M. Searching for Metabolic Pathways of Anaerobic Digestion: A Useful List of the Key Enzymes. In Anaerobic Digestion; Banu, J.R., Ed.; IntechOpen: London, UK, 2018; ISBN 9781838818494. [Google Scholar]
- Quick, J.; Quinlan, A.R.; Loman, N.J. A Reference Bacterial Genome Dataset Generated on the MinIONTM Portable Single-Molecule Nanopore Sequencer. Gigascience 2014, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Salmela, L.; Walve, R.; Rivals, E.; Ukkonen, E. Accurate Self-Correction of Errors in Long Reads Using de Bruijn Graphs. Bioinformatics 2017, 33, 799–806. [Google Scholar] [CrossRef]
- Stoler, N.; Nekrutenko, A. Sequencing Error Profiles of Illumina Sequencing Instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Damme, R.V.; Hölzer, M.; Viehweger, A.; Müller, B.; Bongcam-Rudloff, E.; Brandt, C. Metagenomics Workflow for Hybrid Assembly, Differential Coverage Binning, Metatranscriptomics and Pathway Analysis (MUFFIN). PLoS Comput. Biol. 2021, 17, e1008716. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Yang, Y.; Wang, D.; Cheng, S.H.; Zheng, C.; Zhang, T. Charting the Complexity of the Activated Sludge Microbiome through a Hybrid Sequencing Strategy. Microbiome 2021, 9, 205. [Google Scholar] [CrossRef]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and Benefits of High-Throughput Long-Read Sequencing in Microbial Ecology. Appl. Environ. Microb. 2021, 87, e00626-21. [Google Scholar] [CrossRef]
- Arumugam, K.; Bessarab, I.; Haryono, M.A.S.; Liu, X.; Zuniga-Montanez, R.E.; Roy, S.; Qiu, G.; Drautz-Moses, D.I.; Law, Y.Y.; Wuertz, S.; et al. Analysis Procedures for Assessing Recovery of High Quality, Complete, Closed Genomes from Nanopore Long Read Metagenome Sequencing. BioRxiv 2020. [Google Scholar] [CrossRef]
- Lui, L.M.; Nielsen, T.N.; Arkin, A.P. A Method for Achieving Complete Microbial Genomes and Improving Bins from Metagenomics Data. PLoS Comput. Biol. 2021, 17, e1008972. [Google Scholar] [CrossRef]
- Moss, E.L.; Maghini, D.G.; Bhatt, A.S. Complete, Closed Bacterial Genomes from Microbiomes Using Nanopore Sequencing. Nat. Biotechnol. 2020, 38, 701–707. [Google Scholar] [CrossRef]
- Cerdán, J.M.A.; Tejido-Nuñez, Y.; Aymerich, E.; de GoñiGoñi, J.G.-M.; Garcia-Aguirre, J. A Comprehensive Comparison of Methane and Bio-Based Volatile Fatty Acids Production from Urban and Agro-Industrial Sources. Waste Biomass Valorization 2021, 12, 1357–1369. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online: Http://www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/ (accessed on 11 July 2018).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Popp, D.; Harms, H.; Centler, F. A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion. Microorganisms 2020, 8, 669. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Hartmann, M.; Eriksson, K.M.; Pal, C.; Thorell, K.; Larsson, D.G.J.; Nilsson, R.H. Metaxa2: Improved Identification and Taxonomic Classification of Small and Large Subunit rRNA in Metagenomic Data. Mol. Ecol. Resour. 2015, 15, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, J.; Eriksson, K.M.; Hartmann, M.; Wang, Z.; Shenoy, B.D.; Grelet, G.-A.; Abarenkov, K.; Petri, A.; Rosenblad, M.A.; Nilsson, R.H. Metaxa: A Software Tool for Automated Detection and Discrimination among Ribosomal Small Subunit (12S/16S/18S) Sequences of Archaea, Bacteria, Eukaryotes, Mitochondria, and Chloroplasts in Metagenomes and Environmental Sequencing Datasets. Antonie Van Leeuwenhoek 2011, 100, 471. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. Research in Computational Molecular Biology. In The Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics, Proceedings of the 14th Annual International Conference, RECOMB 2010, Lisbon, Portugal, 25–28 April 2010; Springer: Berlin/Heidelberg, Germany, 2010; pp. 426–440. [Google Scholar]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Tang, Y.-H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An Automated Binning Method to Recover Individual Genomes from Metagenomes Using an Expectation-Maximization Algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EggNOG 4.5: A Hierarchical Orthology Framework with Improved Functional Annotations for Eukaryotic, Prokaryotic and Viral Sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of Nearly 8,000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning Metagenomic Contigs by Coverage and Composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and Translation Initiation Site Prediction in Metagenomic Sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. DRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-Level Functional Profiling of Metagenomes and Metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2019, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Yu, Z.; Zhu, W. Insights into the Populations of Proteolytic and Amino Acid-Fermenting Bacteria from Microbiota Analysis Using In Vitro Enrichment Cultures. Curr. Microbiol. 2018, 75, 1543–1550. [Google Scholar] [CrossRef]
- Örlygsson, J.; Krooneman, J.; Collins, M.D.; Pascual, C.; Gottschal, J.C. Clostridium acetireducens sp. nov., a Novel Amino Acid-Oxidizing, Acetate-Reducing Anaerobic Bacterium. Int. J. Syst. Evol. Microbiol. 1996, 46, 1192. [Google Scholar] [CrossRef]
- Calli, B.; Mertoglu, B.; Inanc, B.; Yenigun, O. Community Changes During Start-up in Methanogenic Bioreactors Exposed to Increasing Levels of Ammonia. Environ. Technol. 2005, 26, 85–91. [Google Scholar] [CrossRef]
- Sprott, G.D.; Patel, G.B. Ammonia Toxicity in Pure Cultures of Methanogenic Bacteria. Syst. Appl. Microbiol. 1986, 7, 358–363. [Google Scholar] [CrossRef]
- Steinhaus, B.; Garcia, M.L.; Shen, A.Q.; Angenent, L.T. A Portable Anaerobic Microbioreactor Reveals Optimum Growth Conditions for the Methanogen Methanosaeta concilii. Appl. Environ. Microb. 2007, 73, 1653–1658. [Google Scholar] [CrossRef]
- Klang, J.; Theuerl, S.; Szewzyk, U.; Huth, M.; Tölle, R.; Klocke, M. Dynamic Variation of the Microbial Community Structure during the Long-time Mono-fermentation of Maize and Sugar Beet Silage. Microb. Biotechnol. 2015, 8, 764–775. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New Insights from the Biogas Microbiome by Comprehensive Genome-Resolved Metagenomics of Nearly 1600 Species Originating from Multiple Anaerobic Digesters. Biotechnol. Biofuels 2020, 13, 25. [Google Scholar] [CrossRef]
- Maus, I.; Klocke, M.; Derenkó, J.; Stolze, Y.; Beckstette, M.; Jost, C.; Wibberg, D.; Blom, J.; Henke, C.; Willenbücher, K.; et al. Impact of Process Temperature and Organic Loading Rate on Cellulolytic/Hydrolytic Biofilm Microbiomes during Biomethanation of Ryegrass Silage Revealed by Genome-Centered Metagenomics and Metatranscriptomics. Environ. Microbiome 2020, 15, 7. [Google Scholar] [CrossRef]
- Pelletier, E.; Kreimeyer, A.; Bocs, S.; Rouy, Z.; Gyapay, G.; Chouari, R.; Rivière, D.; Ganesan, A.; Daegelen, P.; Sghir, A.; et al. “Candidatus Cloacamonas acidaminovorans”: Genome Sequence Reconstruction Provides a First Glimpse of a New Bacterial Division. J. Bacteriol. 2008, 190, 2572–2579. [Google Scholar] [CrossRef]
- Juste-Poinapen, N.M.S.; Turner, M.S.; Rabaey, K.; Virdis, B.; Batstone, D.J. Evaluating the Potential Impact of Proton Carriers on Syntrophic Propionate Oxidation. Sci. Rep. 2015, 5, 18364. [Google Scholar] [CrossRef] [Green Version]
- Ahlert, S.; Zimmermann, R.; Ebling, J.; König, H. Analysis of Propionate-degrading Consortia from Agricultural Biogas Plants. Microbiologyopen 2016, 5, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- De Bok, F.A.M.; Harmsen, H.J.M.; Plugge, C.M.; de Vries, M.C.; Akkermans, A.D.L.; de Vos, W.M.; Stams, A.J.M. The First True Obligately Syntrophic Propionate-Oxidizing Bacterium, Pelotomaculum schinkii sp. nov., Co-Cultured with Methanospirillum hungatei, and Emended Description of the Genus Pelotomaculum. Int. J. Syst. Evol. Microbiol. 2005, 55, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Ueki, A.; Shizuku, T.; Ohtaki, Y.; Ueki, K. Desulfovibrio butyratiphilus sp. nov., a Gram-Negative, Butyrate-Oxidizing, Sulfate-Reducing Bacterium Isolated from an Anaerobic Municipal Sewage Sludge Digester. Int. J. Syst. Evol. Microbiol. 2010, 60, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Schnürer, A.; Westerholm, M. Enrichment and Description of Novel Bacteria Performing Syntrophic Propionate Oxidation at High Ammonia Level. Environ. Microbiol 2021, 23, 1620–1637. [Google Scholar] [CrossRef]
- Moreira, J.P.C.; Diender, M.; Arantes, A.L.; Boeren, S.; Stams, A.J.M.; Alves, M.M.; Alves, J.I.; Sousa, D.Z. Propionate Production from Carbon Monoxide by Synthetic Cocultures of Acetobacterium wieringae and Propionigenic bacteria. Appl. Environ. Microb. 2021, 87, e02839-20. [Google Scholar] [CrossRef]
- Giri, S.; Oña, L.; Waschina, S.; Shitut, S.; Yousif, G.; Kaleta, C.; Kost, C. Metabolic Dissimilarity Determines the Establishment of Cross-Feeding Interactions in Bacteria. Curr. Biol. 2021, 31, 5547–5557.e6. [Google Scholar] [CrossRef]
- Wallrabenstein, C.; Schink, B. Evidence of Reversed Electron Transport in Syntrophic Butyrate or Benzoate Oxidation by Syntrophomonas wolfei and Syntrophus buswellii. Arch. Microbiol. 1994, 162, 136–142. [Google Scholar] [CrossRef]
- Meng, X.; Cao, Q.; Sun, Y.; Huang, S.; Liu, X.; Li, D. 16S rRNA Genes- and Metagenome-Based Confirmation of Syntrophic Butyrate-Oxidizing Methanogenesis Enriched in High Butyrate Loading. Bioresour. Technol. 2022, 345, 126483. [Google Scholar] [CrossRef]
- Solli, L.; Håvelsrud, O.E.; Horn, S.J.; Rike, A.G. A Metagenomic Study of the Microbial Communities in Four Parallel Biogas Reactors. Biotechnol. Biofuels 2014, 7, 146. [Google Scholar] [CrossRef]
- Plugge, C.M.; Balk, M.; Stams, A.J.M. Desulfotomaculum thermobenzoicum subsp. Thermosyntrophicum subsp. nov., a Thermophilic, Syntrophic, Propionate-Oxidizing, Spore-Forming Bacterium. Int. J. Syst. Evol. Microbiol. 2002, 52, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.S.; Ingram-Smith, C. Methanosaeta, the Forgotten Methanogen? Trends Microbiol. 2007, 15, 150–155. [Google Scholar] [CrossRef]
- Rotaru, A.-E.; Shrestha, P.M.; Liu, F.; Shrestha, M.; Shrestha, D.; Embree, M.; Zengler, K.; Wardman, C.; Nevin, K.P.; Lovley, D.R. A New Model for Electron Flow during Anaerobic Digestion: Direct Interspecies Electron Transfer to Methanosaeta for the Reduction of Carbon Dioxide to Methane. Energ. Environ. Sci. 2013, 7, 408–415. [Google Scholar] [CrossRef]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 Long-Read Sequencing Enables the Generation of near-Finished Bacterial Genomes from Pure Cultures and Metagenomes without Short-Read or Reference Polishing. Nat. Methods 2022, 19, 823–826. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| R-1 | R-2 | R-3 | R-4 | R-5 | |
|---|---|---|---|---|---|
| OLR (gCOD/L/d) | 1.55 | 4.65 | |||
| Feeding regime | Continuous feeding over 24 h | 75% daily feed at once, 25% continuously | Continuous feeding over 24 h | 100% daily feed at once | |
| Replicates | -- | Biological | Biological | -- | -- |
| pH | 7.5 ± 0.1 | 7.5 ± 0.1 | 7.5 ± 0.1 | 7.2 ± 0.1 | 7.5 ± 0.2 |
| Total SCFA concentration (mg/L) | 86.9 ± 28.7 | 338.8 ± 413.5 | 56.5 ± 45.2 | 195.6 ± 371.3 | 304.0 ± 455.6 |
| Methane production rate (L/d) | 3.8 ± 0.5 | 4.1 ± 0.4 | 4.4 ± 0.3 | 11.8 ± 2.9 | 10.5 ± 1.9 |
| Microbial biomass (gVS/L) | 0.37 ± 0.01 | 0.34 ± 0.07 | 0.34 ± 0.05 | 0.96 ± 0.15 | 0.79 ± 0.07 |
| Quality | ID | Completeness [%] | Contamination [%] | Length [Mb] | Taxonomic Classification According to GTDB |
|---|---|---|---|---|---|
| HQ | UFZ-5_008 | 100 | 1.69 | 2 | g__58-81 (f_Thermovirgaceae) |
| HQ | UFZ-1_001 | 99.35 | 0.65 | 2.8 | s__Methanothrix soehngenii |
| HQ | UFZ-2_001 | 99.35 | 0.65 | 2.8 | s__Methanothrix soehngenii |
| HQ | UFZ-3_001 | 99.35 | 0.65 | 2.8 | s__Methanothrix soehngenii |
| HQ | UFZ-4_002 | 99.35 | 0.65 | 2.7 | s__Methanothrix soehngenii |
| HQ | UFZ-5_003 | 99.35 | 1.31 | 2.7 | s__Methanothrix soehngenii |
| HQ | UFZ-4_007 | 99.15 | 3.54 | 2 | g__58-81 (f_Thermovirgaceae) |
| MQ | UFZ-5_013 | 97.9 | 6.37 | 4.1 | f__Syntrophobacteraceae |
| HQ | UFZ-1_009 | 97.8 | 0 | 2.2 | g__UBA3900 |
| HQ | UFZ-5_005 | 97.8 | 0 | 2.1 | g__UBA3900 (f_Cloacimonadaceae) |
| HQ | UFZ-1_013 | 97.75 | 1.56 | 1.7 | f__Endomicrobiaceae |
| HQ | UFZ-2_003 | 97.19 | 0.37 | 1.7 | f__Endomicrobiaceae |
| HQ | UFZ-3_003 | 96.68 | 4.39 | 3.7 | s__UBA5314 sp002410325 (f_Syntrophomonadaceae) |
| HQ | UFZ-5_009 | 96.07 | 0.44 | 1.6 | f__Endomicrobiaceae |
| MQ | UFZ-4_004 | 95.82 | 5.82 | 3.4 | s__UBA5314 sp002410325 (f_Syntrophomonadaceae) |
| HQ | UFZ-5_004 | 95.42 | 0.65 | 3.8 | s__Methanosarcina mazei |
| MQ | UFZ-2_004 | 95.15 | 6.13 | 3.8 | s__UBA5314 sp002410325 (f_Syntrophomonadaceae) |
| HQ | UFZ-3_007 | 94.76 | 1.85 | 1.7 | g__58-81 (f_Thermovirgaceae) |
| HQ | UFZ-2_002 | 94.56 | 1.72 | 2.7 | g__Methanoculleus |
| HQ | UFZ-4_001 | 94.44 | 2.75 | 2.7 | g__Methanoculleus |
| HQ | UFZ-4_008 | 94.25 | 1.79 | 2.4 | s__UBA2256 sp001603165 (o_Treponematales) |
| HQ | UFZ-5_007 | 94.25 | 3.58 | 2.5 | s__UBA2256 sp001603165 (o_Treponematales) |
| HQ | UFZ-5_010 | 93.47 | 3.14 | 2.5 | f__4484-276 (o_Bacteroidales) |
| HQ | UFZ-1_010 | 93.37 | 1.09 | 2.6 | f__Syntrophomonadaceae |
| HQ | UFZ-1_014 | 93.1 | 2.98 | 2.4 | s__UBA2256 sp001603165 (o_Treponematales) |
| HQ | UFZ-1_018 | 93.04 | 4.25 | 4 | f__Syntrophobacteraceae |
| HQ | UFZ-4_011 | 92.42 | 2.32 | 4 | f__Syntrophobacteraceae |
| HQ | UFZ-5_002 | 92.19 | 3.16 | 2.4 | g__Methanoculleus |
| HQ | UFZ-3_004 | 92.17 | 4.4 | 2.5 | f__4484-276 (o_Bacteroidales) |
| HQ | UFZ-1_004 | 91.73 | 0.21 | 2.5 | s__Syntrophomonas wolfei |
| MQ | UFZ-3_002 | 89.58 | 5.28 | 2.7 | g__Methanoculleus |
| MQ | UFZ-1_019 | 87.85 | 2.59 | 2.7 | s__Mesotoga sp002305955 |
| MQ | UFZ-1_003 | 86.8 | 2.69 | 2.1 | g__Methanoculleus |
| MQ | UFZ-1_020 | 86.69 | 3.12 | 2.7 | s__UBA5314 sp002410325 (f_Syntrophomonadaceae) |
| MQ | UFZ-3_005 | 86.67 | 1.11 | 2.8 | f__BBW3 (o_Bacteroidales) |
| MQ | UFZ-1_006 | 86.27 | 0.54 | 2.6 | f__4484-276 (o_Bacteroidales) |
| MQ | UFZ-3_009 | 86.13 | 9.59 | 3.7 | f__Syntrophobacteraceae |
| MQ | UFZ-5_018 | 83.06 | 2.72 | 1.9 | g__Syntrophomonas_B |
| MQ | UFZ-4_003 | 80.88 | 0.47 | 2.3 | s__Mesotoga infera_B |
| MQ | UFZ-2_008 | 72.42 | 1.61 | 2.2 | f__BBW3 (o_Bacteroidales) |
| MQ | UFZ-5_012 | 71.28 | 1.19 | 2 | g__Corynebacterium |
| MQ | UFZ-5_014 | 68.95 | 2.01 | 1.3 | g__Methanospirillum |
| MQ | UFZ-5_017 | 68.02 | 3.11 | 1.8 | g__SR-FBR-E99 (o_Bacteroidales) |
| MQ | UFZ-4_010 | 67.63 | 0.86 | 1.7 | f__4484-276 (o_Bacteroidales) |
| MQ | UFZ-4_006 | 67.58 | 4.4 | 1.3 | s__Cloacimonas acidaminovorans |
| MQ | UFZ-5_015 | 67.1 | 3.22 | 1.2 | f__DTU023 (o_Saccharofermentanales) |
| MQ | UFZ-2_006 | 63.94 | 1.34 | 1.7 | f__4484-276 (o_Bacteroidales) |
| MQ | UFZ-2_005 | 62.8 | 1.12 | 0.4 | s__UBA2558 sp002340425 (o_Paceibacterales) |
| MQ | UFZ-4_012 | 62.67 | 1.21 | 1.4 | p__Firmicutes_A |
| MQ | UFZ-3_008 | 60.22 | 2.88 | 0.9 | f__Endomicrobiaceae |
| MQ | UFZ-5_019 | 56.14 | 5.65 | 1.4 | f__Salinivirgaceae |
| MQ | UFZ-2_007 | 55.22 | 1.66 | 1.3 | g__Methanoculleus |
| MQ | UFZ-1_029 | 55.05 | 2 | 0.9 | g__58-81 (f_Thermovirgaceae) |
| MQ | UFZ-5_006 | 53.45 | 0 | 1.6 | - |
| MQ | UFZ-2_010 | 50.76 | 2.01 | 1 | g__UBA3900 (f_Cloacimonadaceae) |
| MQ | UFZ-2_009 | 50.08 | 0.22 | 2.1 | f__Syntrophobacteraceae |
| Quality | ID | Completeness [%] | Contamination [%] | Length [Mb] | GTDB Classification | TYGS Classification * |
|---|---|---|---|---|---|---|
| HQ | UFZ-4H1 | 100 | 1.1 | 2.2 | s__Cloacimonas acidaminovorans | Potential new species |
| HQ | UFZ-4H6 | 99.84 | 0.47 | 3.0 | s__Mesotoga infera_B | Mesotoga infera |
| HQ | UFZ-4H12 | 99.35 | 0.65 | 2.9 | s__Methanothrix soehngenii | Methanothrix soehngenii |
| HQ | UFZ-1H11 | 98.98 | 1.86 | 3.2 | g__DTU019 (f_Syntrophomonadaceae) | Potential new species |
| HQ | UFZ-4H3 | 98.87 | 2.1 | 4.4 | f__Syntrophobacteraceae | Potential new species |
| HQ | UFZ-4H4 | 98.09 | 4.67 | 4.4 | s__UBA5314 sp002410325 (f_Syntrophobacteraceae) | Desulfovibrio paquesii |
| HQ | UFZ-1H7 | 97.86 | 1.23 | 3.0 | s__Syntrophomonas wolfei | Syntrophomonas wolfei |
| HQ | UFZ-1H3 | 97.22 | 0.65 | 2.9 | g__Methanoculleus | Potential new species |
| HQ | UFZ-4H14 | 96.55 | 0 | 2.7 | s__UBA2256 sp001603165 (o_Treponematales) | Potential new species |
| HQ | UFZ-4H10 | 95.84 | 1.69 | 2.2 | g__58-81 (f_Thermovirgaceae) | Potential new species |
| MQ | UFZ-4H11 | 91.99 | 7.01 | 3.4 | s__Syntrophomonas wolfei | Potential new species |
| HQ | UFZ-1H10 | 90.59 | 0.54 | 2.7 | f__4484-476 (o_Bacteroidales) | Potential new species |
| MQ | UFZ-1H9 | 78.19 | 0.58 | 1.8 | g__Syntrophomonas_B | Potential new species |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, D.; Popp, D.; Bonk, F.; Kleinsteuber, S.; Harms, H.; Centler, F. Metagenomic Analysis of Anaerobic Microbial Communities Degrading Short-Chain Fatty Acids as Sole Carbon Sources. Microorganisms 2023, 11, 420. https://doi.org/10.3390/microorganisms11020420
Becker D, Popp D, Bonk F, Kleinsteuber S, Harms H, Centler F. Metagenomic Analysis of Anaerobic Microbial Communities Degrading Short-Chain Fatty Acids as Sole Carbon Sources. Microorganisms. 2023; 11(2):420. https://doi.org/10.3390/microorganisms11020420
Chicago/Turabian StyleBecker, Daniela, Denny Popp, Fabian Bonk, Sabine Kleinsteuber, Hauke Harms, and Florian Centler. 2023. "Metagenomic Analysis of Anaerobic Microbial Communities Degrading Short-Chain Fatty Acids as Sole Carbon Sources" Microorganisms 11, no. 2: 420. https://doi.org/10.3390/microorganisms11020420