Cryo-EM Structure and Molecular Dynamics Analysis of the Fluoroquinolone Resistant Mutant of the AcrB Transporter from Salmonella

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of AcrB G288D
2.2. Protein Overexpression and Purification
2.3. Negative Stain Electron Microscopy
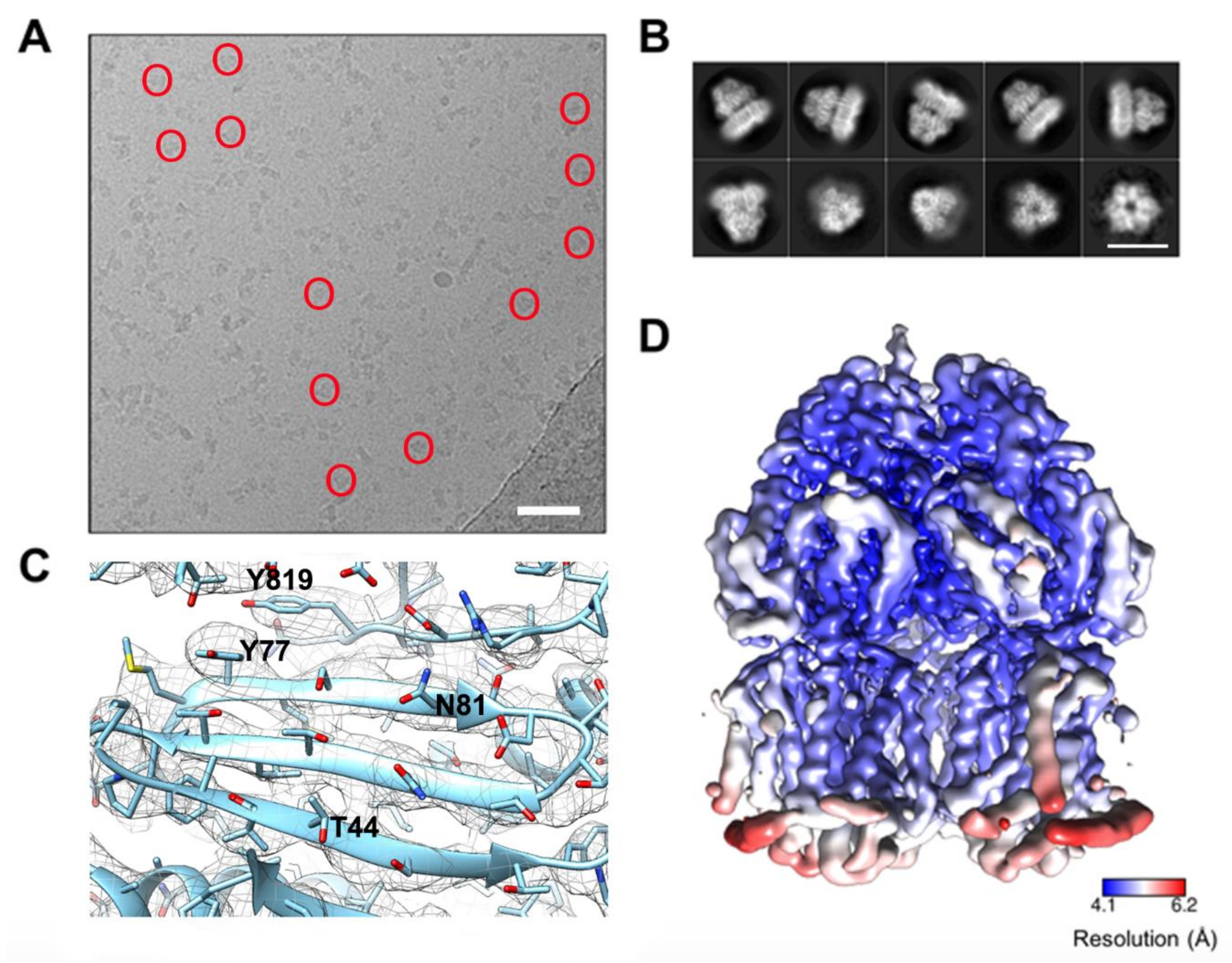
2.4. Cryo-Electron Microscopy
2.5. Methods for Structural Analysis
2.6. Homology Modelling
2.7. Molecular Dynamics Simulations
2.8. Post-Processing of MD Trajectories
3. Results
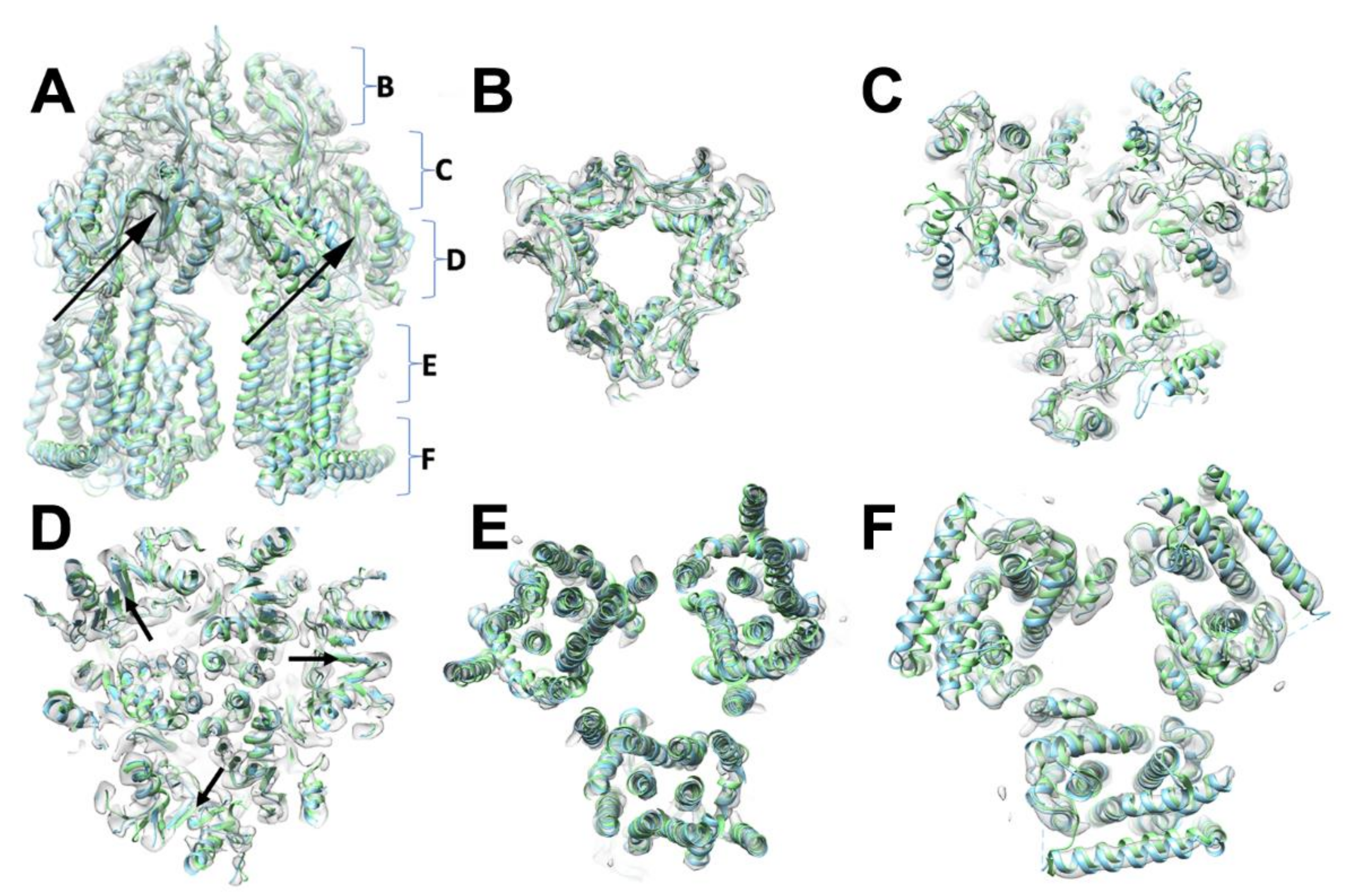
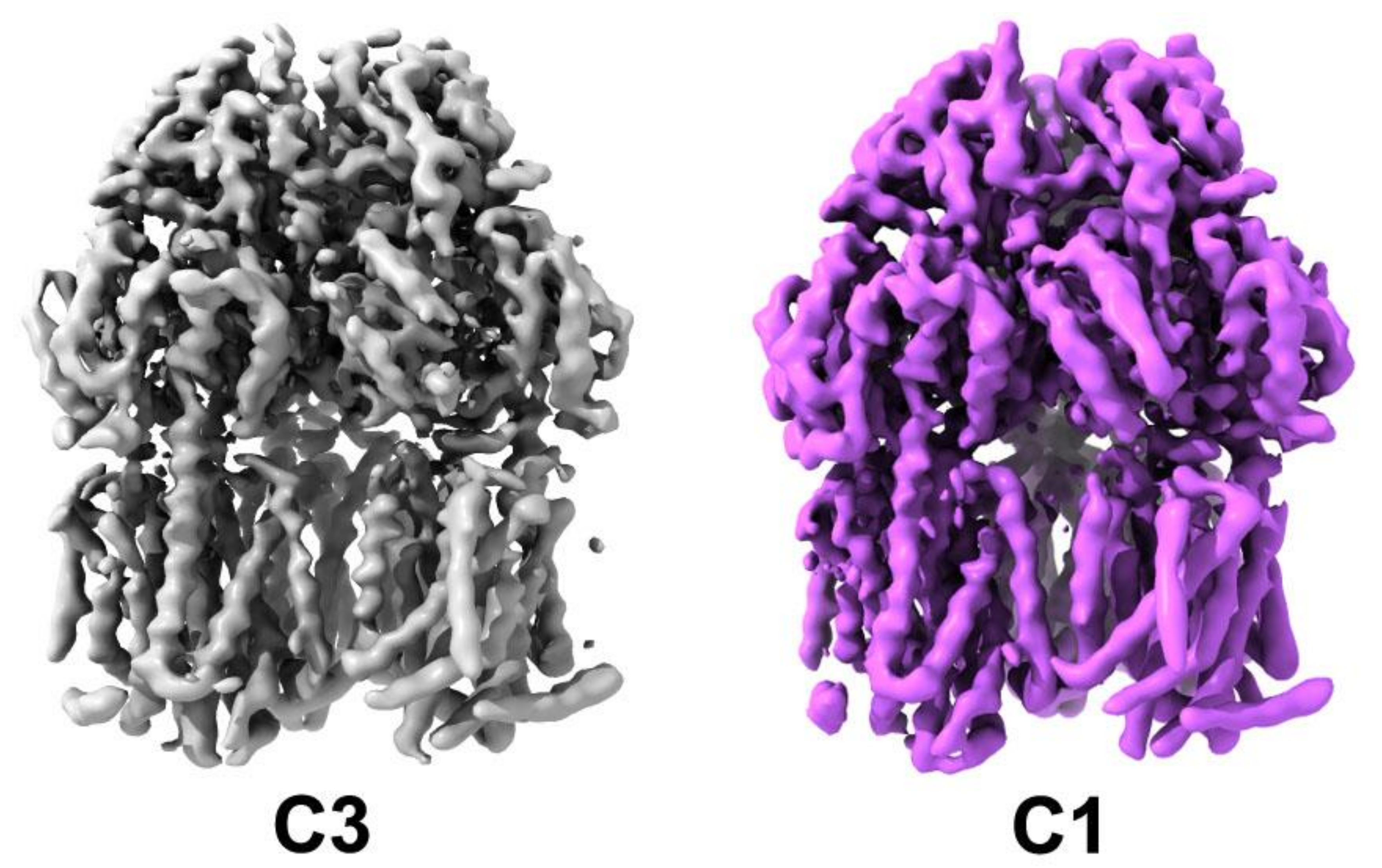
3.1. Oligomeric State and Overall Fold of S. Typhimurium AcrB
3.2. Comparison between Salmonella AcrB G288D Processed in the C3 Space Group and the WT E. coli Transporters
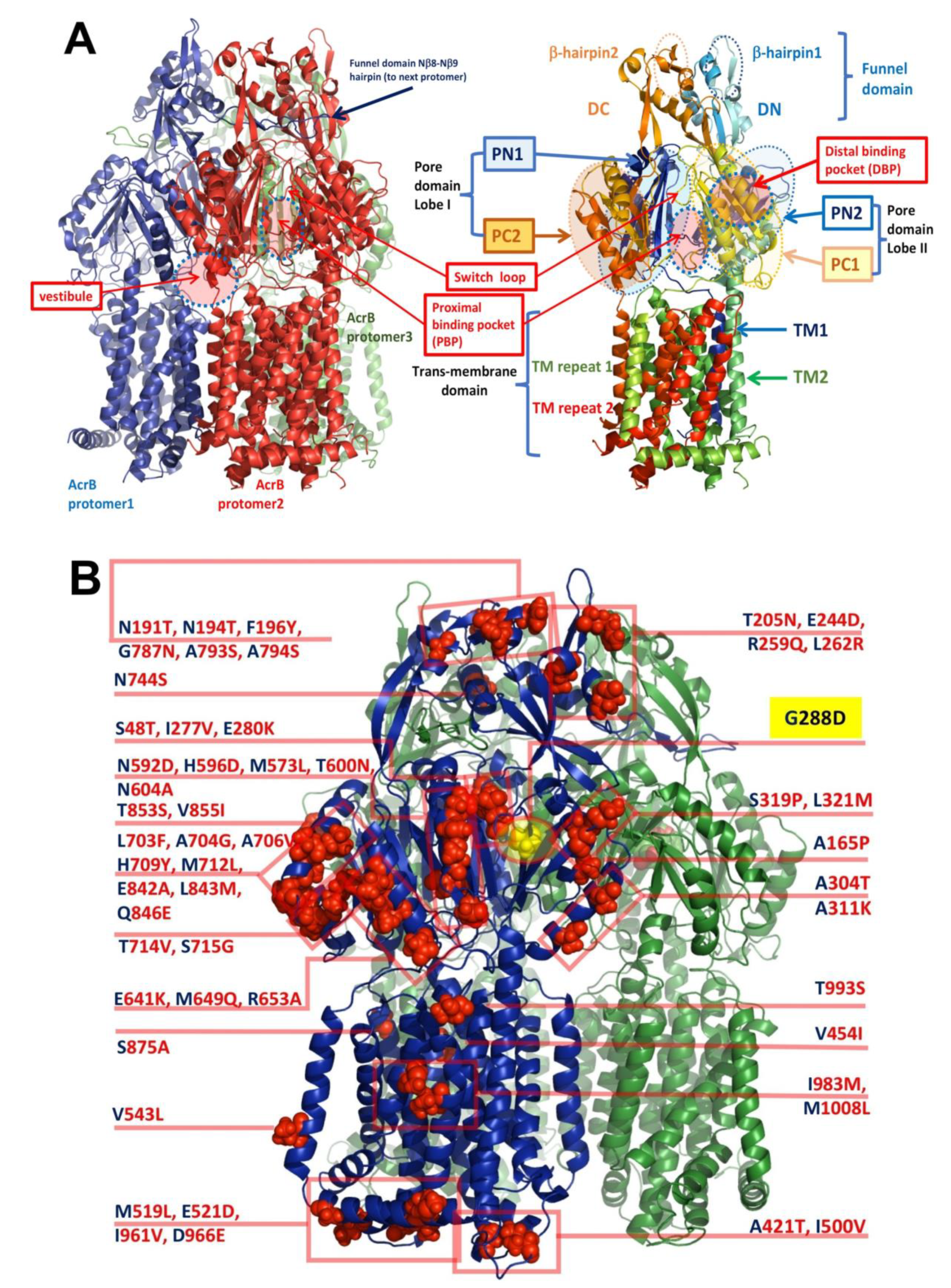
3.3. Implications for Drug Selectivity and Efflux Efficiency
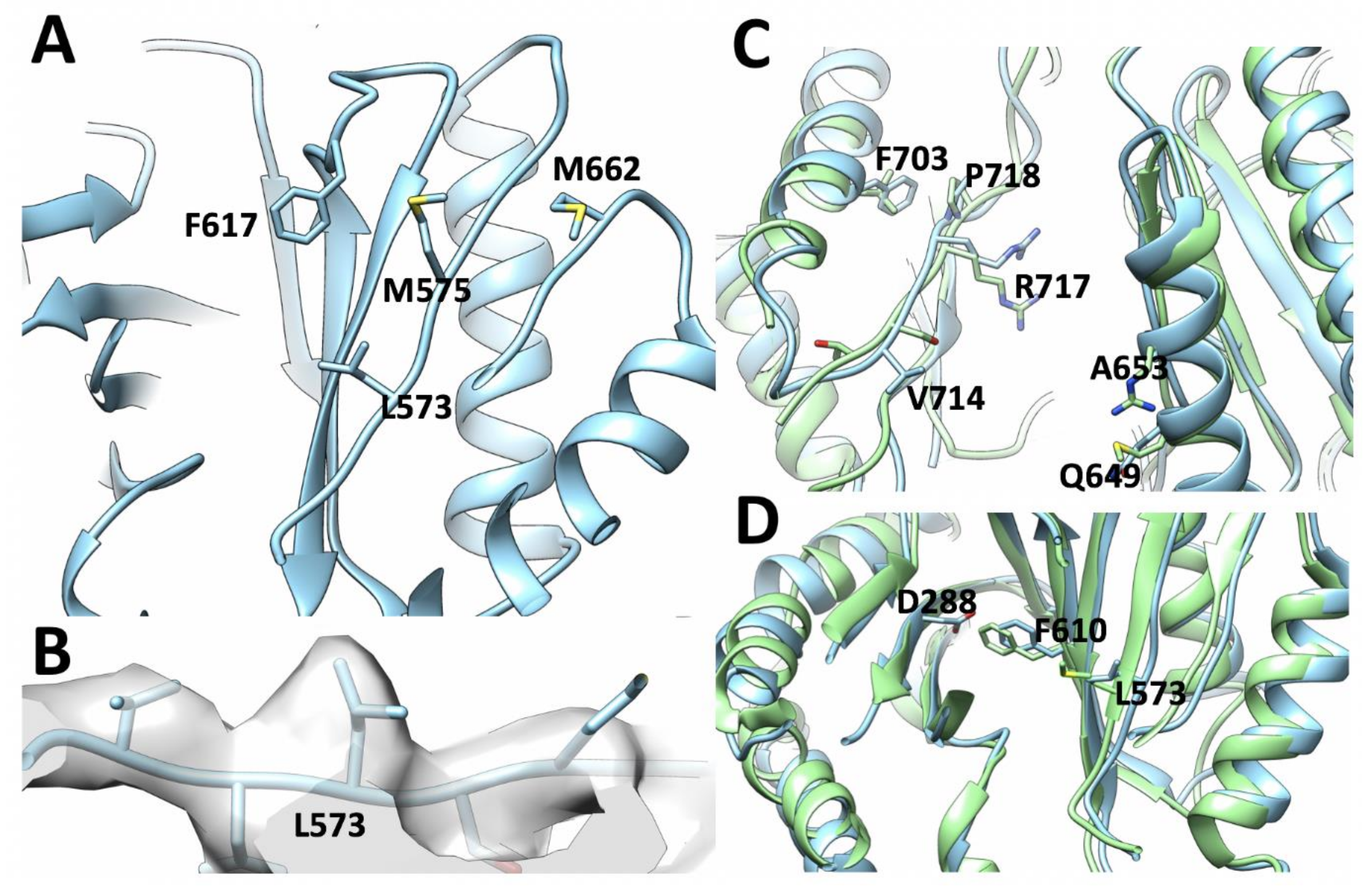
3.4. Structural Changes Attributable to G288D Mutation
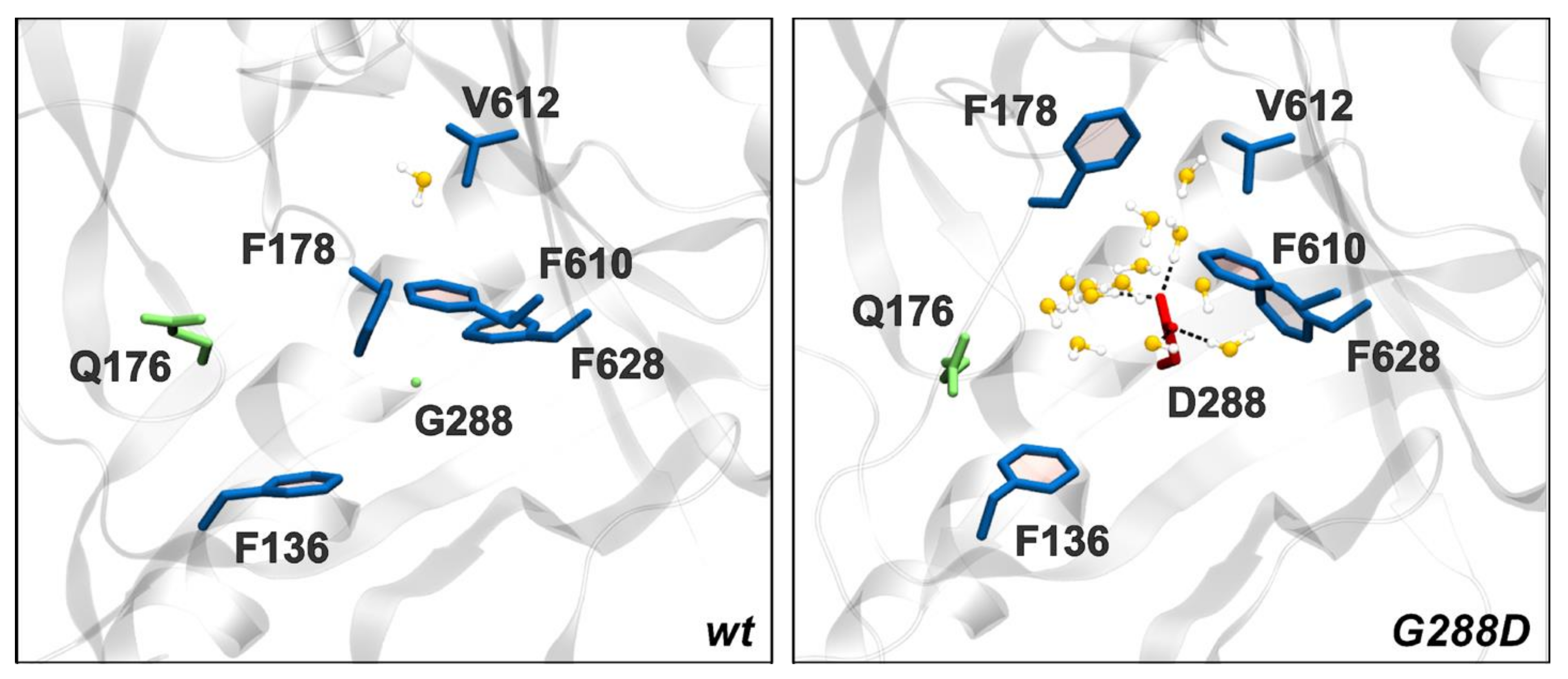
3.5. Mechanism of G288D Mutation Inferred from MD Simulations
4. Discussion
On the DBP Pocket and the Effect of G288D
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Curiao, T.; Marchi, E.; Grandgirard, D.; León-Sampedro, R.; Viti, C.; Leib, S.L.; Baquero, F.; Oggioni, M.R.; Martinez, J.L.; Coque, T.M. Multiple adaptive routes of Salmonella enterica Typhimurium to biocide and antibiotic exposure. BMC Genomics 2016, 17, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, R.; Bavro, V.N. Assembly and transport mechanism of tripartite drug efflux systems. Biochim. Biophys. Acta 2009, 1794, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Kobylka, J.; Kuth, M.S.; Müller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A mean, keen, drug efflux machine. Ann. N. Y. Acad. Sci. 2019, 1459, 38–68. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Latifi, T.; Groisman, E.A. Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2006, 59, 126–141. [Google Scholar] [CrossRef]
- Janganan, T.K.; Bavro, V.N.; Zhang, L.; Matak-Vinkovic, D.; Barrera, N.P.; Venien-Bryan, C.; Robinson, C.V.; Borges-Walmsley, M.I.; Walmsley, A.R. Evidence for the assembly of a bacterial tripartite multidrug pump with a stoichiometry of 3:6:3. J. Biol. Chem. 2011, 286, 26900–26912. [Google Scholar] [CrossRef] [Green Version]
- Symmons, M.F.; Marshall, R.L.; Bavro, V.N. Architecture and roles of periplasmic adaptor proteins in tripartite efflux assemblies. Front. Microbiol. 2015, 6, 513. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 2017, 6, e24905. [Google Scholar] [CrossRef]
- McNeil, H.E.; Alav, I.; Torres, R.C.; Rossiter, A.E.; Laycock, E.; Legood, S.; Kaur, I.; Davies, M.; Wand, M.; Webber, M.A.; et al. Identification of binding residues between periplasmic adapter protein (PAP) and RND efflux pumps explains PAP-pump promiscuity and roles in antimicrobial resistance. PLoS Pathog. 2019, 15, e1008101. [Google Scholar] [CrossRef]
- Horiyama, T.; Yamaguchi, A.; Nishino, K. TolC dependency of multidrug efflux systems in Salmonella enterica serovar Typhimurium. J. Antimicrob. Chemother. 2010, 65, 1372–1376. [Google Scholar] [CrossRef]
- Wang-Kan, X.; Blair, J.M.A.; Chirullo, B.; Betts, J.; La Ragione, R.M.; Ivens, A.; Ricci, V.; Opperman, T.J.; Piddock, L.J.V. Lack of AcrB Efflux Function Confers Loss of Virulence on Salmonella enterica Serovar Typhimurium. MBio 2017, 8, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, J.M.A.; Smith, H.E.; Ricci, V.; Lawler, A.J.; Thompson, L.J.; Piddock, L.J. V Expression of homologous RND efflux pump genes is dependent upon AcrB expression: Implications for efflux and virulence inhibitor design. J. Antimicrob. Chemother. 2015, 70, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Piddock, L.J.V. Ciprofloxacin selects for multidrug resistance in Salmonella enterica serovar Typhimurium mediated by at least two different pathways. J. Antimicrob. Chemother. 2009, 63, 909–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, S.; Phillips, C.R.; Ekanayaka, A.S.; Piddock, L.J.V.; Webber, M.A. Inhibition of multidrug efflux as a strategy to prevent biofilm formation. J. Antimicrob. Chemother. 2014, 69, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef]
- Seeger, M.A.; Schiefner, A.; Eicher, T.; Verrey, F.; Diederichs, K.; Pos, K.M. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 2006, 313, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Seeger, M.A.; von Ballmoos, C.; Eicher, T.; Brandstätter, L.; Verrey, F.; Diederichs, K.; Pos, K.M. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat. Struct. Mol. Biol. 2008, 15, 199–205. [Google Scholar] [CrossRef]
- Eicher, T.; Cha, H.; Seeger, M.A.; Brandstatter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grutter, M.G.; Diederichs, K.; et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef] [Green Version]
- Takatsuka, Y.; Nikaido, H. Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J. Bacteriol. 2009, 191, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, Y.; Yamane, T.; Terada, T.; Moritsugu, K.; Fujisaki, H.; Murakami, S.; Ikeguchi, M.; Kidera, A. Energetics and conformational pathways of functional rotation in the multidrug transporter AcrB. Elife 2018, 7, e31715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargiu, A.V.; Ramaswamy, V.K.; Malvacio, I.; Malloci, G.; Kleinekathöfer, U.; Ruggerone, P. Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump. Biochim. Biophys. Acta -Gen. Subj. 2018, 1862, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Husain, F.; Nikaido, H. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 2010, 78, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Tam, H.-K.; Malviya, V.N.; Foong, W.-E.; Herrmann, A.; Malloci, G.; Ruggerone, P.; Vargiu, A.V.; Pos, K.M. Binding and Transport of Carboxylated Drugs by the Multidrug Transporter AcrB. J. Mol. Biol. 2019, 2019 432, 861–877. [Google Scholar] [CrossRef]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Hayashi, K.; Nagata, C.; Hoshino, K.; Onodera, Y.; Nishino, K.; Yamaguchi, A. Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013, 500, 102–106. [Google Scholar] [CrossRef]
- Zwama, M.; Yamasaki, S.; Nakashima, R.; Sakurai, K.; Nishino, K.; Yamaguchi, A. Multiple entry pathways within the efflux transporter AcrB contribute to multidrug recognition. Nat. Commun. 2018, 9, 124. [Google Scholar] [CrossRef]
- Oswald, C.; Tam, H.-K.; Pos, K.M. Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB. Nat. Commun. 2016, 7, 13819. [Google Scholar] [CrossRef]
- Eicher, T.; Seeger, M.A.; Anselmi, C.; Zhou, W.; Brandstätter, L.; Verrey, F.; Diederichs, K.; Faraldo-Gómez, J.D.; Pos, K.M. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 2014, 3, e03145. [Google Scholar] [CrossRef] [Green Version]
- Atac, N.; Kurt-Azap, O.; Dolapci, I.; Yesilkaya, A.; Ergonul, O.; Gonen, M.; Can, F. The Role of AcrAB-TolC Efflux Pumps on Quinolone Resistance of E. coli ST131. Curr. Microbiol. 2018, 75, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Haycocks, J.R.J.; Middlemiss, A.D.; Kettles, R.A.; Sellars, L.E.; Ricci, V.; Piddock, L.J.V.; Grainger, D.C. The multiple antibiotic resistance operon of enteric bacteria controls DNA repair and outer membrane integrity. Nat. Commun. 2017, 8, 1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, N.; Sharma, P.; Ricci, V.; Piddock, L.J.V. Regulation of the AcrAB-TolC efflux pump in Enterobacteriaceae. Res. Microbiol. 2018, 169, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Bavro, V.N.; Ricci, V.; Modi, N.; Cacciotto, P.; Kleinekathӧfer, U.; Ruggerone, P.; Vargiu, A.V.; Baylay, A.J.; Smith, H.E.; et al. AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc. Natl. Acad. Sci. USA 2015, 112, 3511–3516. [Google Scholar] [CrossRef] [Green Version]
- Piddock, L.J.; Griggs, D.J.; Hall, M.C.; Jin, Y.F. Ciprofloxacin resistance in clinical isolates of Salmonella typhimurium obtained from two patients. Antimicrob. Agents Chemother. 1993, 37, 662–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piddock, L.J.; Whale, K.; Wise, R. Quinolone resistance in salmonella: Clinical experience. Lancet (London, UK) 1990, 335, 1459. [Google Scholar] [CrossRef]
- Parmar, M.; Rawson, S.; Scarff, C.A.; Goldman, A.; Dafforn, T.R.; Muench, S.P.; Postis, V.L.G. Using a SMALP platform to determine a sub-nm single particle cryo-EM membrane protein structure. Biochim. Biophys. Acta. Biomembr. 2018, 1860, 378–383. [Google Scholar] [CrossRef]
- Lee, S.C.; Knowles, T.J.; Postis, V.L.G.; Jamshad, M.; Parslow, R.A.; Lin, Y.-P.; Goldman, A.; Sridhar, P.; Overduin, M.; Muench, S.P.; et al. A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat. Protoc. 2016, 11, 1149–1162. [Google Scholar] [CrossRef]
- Roach, P.C.J.; Postis, V.L.G.; Deacon, S.E.; Wright, G.S.A.; Ingram, J.C.; Xia, X.; McPherson, M.J.; Baldwin, S.A. Large-scale preparation of bacterial cell membranes by tangential flow filtration. Mol. Membr. Biol. 2008, 25, 609–616. [Google Scholar] [CrossRef]
- Postis, V.; Rawson, S.; Mitchell, J.K.; Lee, S.C.; Parslow, R.A.; Dafforn, T.R.; Baldwin, S.A.; Muench, S.P. The use of SMALPs as a novel membrane protein scaffold for structure study by negative stain electron microscopy. Biochim. Biophys. Acta 2015, 1848, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimanius, D.; Forsberg, B.O.; Scheres, S.H.; Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 2016, 5, 18722. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanal, A.; Lohkamp, B.; Emsley, P. Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Sci. 2020, 29, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Sennhauser, G.; Amstutz, P.; Briand, C.; Storchenegger, O.; Grütter, M.G. Drug Export Pathway of Multidrug Exporter AcrB Revealed by DARPin Inhibitors. PLoS Biol. 2006, 5, e7. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar]
- Topf, M.; Lasker, K.; Webb, B.; Wolfson, H.; Chiu, W.; Sali, A. Protein structure fitting and refinement guided by cryo-EM density. Structure 2008, 16, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargiu, A.V.; Nikaido, H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 20637–20642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Huang, Y.; Walker, R.C.; Lin, C.; Cheatham, T.E., III; Mermelstein, D.J.; Simmerling, C.; Li, P.; Roitberg, A.; Onufriev, A.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Vargiu, A.V.; Ruggerone, P.; Opperman, T.J.; Nguyen, S.T.; Nikaido, H. Molecular Mechanism of MBX2319 Inhibition of Escherichia coli AcrB Multidrug Efflux Pump and Comparison with Other Inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6224–6234. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.K.; Vargiu, A.V.; Malloci, G.; Dreier, J.; Ruggerone, P. Molecular Rationale behind the Differential Substrate Specificity of Bacterial RND Multi-Drug Transporters. Sci. Rep. 2017, 7, 8075. [Google Scholar] [CrossRef] [Green Version]
- Sjuts, H.; Vargiu, A.V.; Kwasny, S.M.; Nguyen, S.T.; Kim, H.-S.; Ding, X.; Ornik, A.R.; Ruggerone, P.; Bowlin, T.L.; Nikaido, H.; et al. Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, 3509–3514. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Das, D.; Xu, Q.S.; Lee, J.Y.; Ankoudinova, I.; Huang, C.; Lou, Y.; DeGiovanni, A.; Kim, R.; Kim, S.-H. Crystal structure of the multidrug efflux transporter AcrB at 3.1Å resolution reveals the N-terminal region with conserved amino acids. J. Struct. Biol. 2007, 158, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Fu, Z.; Xu, G.G.; Grassucci, R.A.; Zhang, Y.; Frank, J.; Hendrickson, W.A.; Guo, Y. Structure and activity of lipid bilayer within a membrane-protein transporter. Proc. Natl. Acad. Sci. USA 2018, 115, 12985–12990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, J.D.; de Oliveira, C.A.F.; McCammon, J.A. POVME: An algorithm for measuring binding-pocket volumes. J. Mol. Graph. Model. 2011, 29, 773–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 1991, 12, 337–357. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.-W.; Kim, H.-B.; Murakami, S.; Gupta, G.; Kim, C.-Y.; Terwilliger, T.C. Crystal structure of AcrB complexed with linezolid at 3.5 Å resolution. J. Struct. Funct. Genomics 2013, 14, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R.T.; Travers, T.; Cha, H.-J.; Phillips, J.L.; Gnanakaran, S.; Pos, K.M. Switch Loop Flexibility Affects Substrate Transport of the AcrB Efflux Pump. J. Mol. Biol. 2017, 429, 3863–3874. [Google Scholar] [CrossRef] [PubMed]
- Hryc, C.F.; Chen, D.-H.; Afonine, P.V.; Jakana, J.; Wang, Z.; Haase-Pettingell, C.; Jiang, W.; Adams, P.D.; King, J.A.; Schmid, M.F.; et al. Accurate model annotation of a near-atomic resolution cryo-EM map. Proc. Natl. Acad. Sci. USA 2017, 114, 3103–3108. [Google Scholar] [CrossRef] [Green Version]
- Soparkar, K.; Kinana, A.D.; Weeks, J.W.; Morrison, K.D.; Nikaido, H.; Misra, R. Reversal of the Drug Binding Pocket Defects of the AcrB Multidrug Efflux Pump Protein of Escherichia coli. J. Bacteriol. 2015, 197, 3255–3264. [Google Scholar] [CrossRef] [Green Version]
- Vargiu, A.V.; Ramaswamy, V.K.; Malloci, G.; Malvacio, I.; Atzori, A.; Ruggerone, P. Computer simulations of the activity of RND efflux pumps. Res. Microbiol. 2018, 169, 384–392. [Google Scholar] [CrossRef]
- Psakis, G.; Polaczek, J.; Essen, L.-O. AcrB et al.: Obstinate contaminants in a picogram scale. One more bottleneck in the membrane protein structure pipeline. J. Struct. Biol. 2009, 166, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Glover, C.A.P.; Postis, V.L.G.; Charalambous, K.; Tzokov, S.B.; Booth, W.I.; Deacon, S.E.; Wallace, B.A.; Baldwin, S.A.; Bullough, P.A. AcrB contamination in 2-D crystallization of membrane proteins: Lessons from a sodium channel and a putative monovalent cation/proton antiporter. J. Struct. Biol. 2011, 176, 419–424. [Google Scholar] [CrossRef]
- Su, C.-C.; Morgan, C.E.; Kambakam, S.; Rajavel, M.; Scott, H.; Huang, W.; Emerson, C.C.; Taylor, D.J.; Stewart, P.L.; Bonomo, R.A.; et al. Cryo-Electron Microscopy Structure of an Acinetobacter baumannii Multidrug Efflux Pump. MBio 2019, 10, e01295-e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trampari, E.; Holden, E.R.; Wickham, G.J.; Ravi, A.; Prischi, F.; de Oliveira Martins, L.; Savva, G.M.; Bavro, V.N.; Webber, M.A. Antibiotics select for novel pathways of resistance in biofilms. bioRxiv 2019, 605212. [Google Scholar] [CrossRef]
- Yao, H.; Shen, Z.; Wang, Y.; Deng, F.; Liu, D.; Naren, G.; Dai, L.; Su, C.-C.; Wang, B.; Wang, S.; et al. Emergence of a Potent Multidrug Efflux Pump Variant That Enhances Campylobacter Resistance to Multiple Antibiotics. MBio 2016, 7, e01543-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnert, J.A.; Schuster, S.; Fähnrich, E.; Trittler, R.; Kern, W. V Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J. Antimicrob. Chemother. 2007, 59, 1216–1222. [Google Scholar] [CrossRef]
- Ramaswamy, V.K.; Vargiu, A.V.; Malloci, G.; Dreier, J.; Ruggerone, P. Molecular Determinants of the Promiscuity of MexB and MexY Multidrug Transporters of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 1144. [Google Scholar] [CrossRef]
- Schuster, S.; Kohler, S.; Buck, A.; Dambacher, C.; König, A.; Bohnert, J.A.; Kern, W. V Random mutagenesis of the multidrug transporter AcrB from Escherichia coli for identification of putative target residues of efflux pump inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6870–6878. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, J.A.; Schuster, S.; Seeger, M.A.; Fähnrich, E.; Pos, K.M.; Kern, W. V Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 2008, 190, 8225–8229. [Google Scholar] [CrossRef] [Green Version]
- Reading, E.; Ahdash, Z.; Fais, C.; Ricci, V.; Kan, X.W.; Grimsey, E.; Stone, J.; Malloci, G.; Lau, A.M.; Findlay, H.; et al. Perturbed structural dynamics underlie inhibition and altered specificity of the multidrug efflux pump AcrB. bioRxiv 2020, 063511. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homology Model | Chain | RMSD (Reference Structure: EcAcrB 4DX7) | ||
|---|---|---|---|---|
| L-Conformer | T-Conformer | O-Conformer | ||
| 1 (template: 2J8S) | A | 2.1 (0.1) | 2.7 (0.1) | 3.2 (0.1) |
| B | 3.0 (0.1) | 2.3 (0.1) | 4.1 (0.1) | |
| C | 3.5 (0.1) | 3.9 (0.1) | 2.2 (0.1) | |
| 2 (template: 4DX5) | A | 2.4 (0.1) | 3.1 (0.1) | 3.0 (0.1) |
| B | 3.0 (0.1) | 2.2 (0.1) | 3.9 (0.1) | |
| C | 3.2 (0.1) | 3.9 (0.1) | 2.2 (0.1) | |
| 3 (template: 4DX7) | A | 2.1 (0.1) | 2.8 (0.1) | 3.4 (0.1) |
| B | 3.1 (0.1) | 2.3 (0.1) | 4.1 (0.1) | |
| C | 3.1 (0.1) | 3.5 (0.1) | 2.1 (0.1) | |
| System | Volume of DBP [Å3] | ||
|---|---|---|---|
| L-Conformer | T-Conformer | O-Conformer | |
| EcAcrBWT1 | 763 (86) | 2315 (76) | 1094 (76) |
| STmAcrBWT2 | 957 (93) | 1534 (163) | 991 (96) |
| STmAcrBG288D2 | 807 (78) | 1979 (72) | 1151 (64) |
| System | Radius of Gyration [Å] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| L-Conformer | T-Conformer | O-Conformer | |||||||
| Whole | Upper | HP Trap | Whole | Upper | HP Trap | Whole | Upper | HP Trap | |
| EcAcrBWT1 | 10.2 (0.1) | 9.0 (0.1) | 6.4 (0.3) | 10.8 (0.1) | 9.4 (0.1) | 7.1 (0.3) | 10.6 (0.1) | 9.9 (0.1) | 6.2 (0.0) |
| STmAcrBWT2 | 10.5 (0.1) | 9.1 (0.1) | 6.3 (0.2) | 10.7 (0.2) | 8.7 (0.1) | 6.9 (0.2) | 10.9 (0.1) | 9.8 (0.1) | 6.3 (0.2) |
| STmAcrBG288D2 | 10.5 (0.1) | 9.2 (0.1) | 6.4 (0.1) | 11.2 (0.1) | 9.2 (0.2) | 8.0 (0.2) | 11.0 (0.1) | 9.9 (0.1) | 6.5 (0.2) |
| System | AcrB Conformer | ||
|---|---|---|---|
| L-Conformer | T-Conformer | O-Conformer | |
| # 1st solv. shell waters | |||
| STmAcrBWT | - | 0.1 (0.3) | 0.0 (0.1) |
| STmAcrBG288D | - | 6.3 (0.6) | 3.1 (0.3) |
| # 2nd solv. shell waters | |||
| STmAcrBWT | 0.0 (0.1) | 0.5 (0.4) | 0.2 (0.2) |
| STmAcrBG288D | 0.2 (0.3) | 11.3 (1.2) | 5.7 (0.5) |
| System | Number of Contacts (PC1-PC2) | ||
|---|---|---|---|
| L-Conformer | T-Conformer | O-Conformer | |
| EcAcrBWT1 | 9 (5) | 10 (2) | 38 (2) |
| STmAcrBWT2 | 12 (4) | 6 (2) | 38 (7) |
| STmAcrBG288D2 | 1(1) | 7(2) | 31 (7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.M.; Fais, C.; Parmar, M.; Cheruvara, H.; Marshall, R.L.; Hesketh, S.J.; Feasey, M.C.; Ruggerone, P.; Vargiu, A.V.; Postis, V.L.G.; et al. Cryo-EM Structure and Molecular Dynamics Analysis of the Fluoroquinolone Resistant Mutant of the AcrB Transporter from Salmonella. Microorganisms 2020, 8, 943. https://doi.org/10.3390/microorganisms8060943
Johnson RM, Fais C, Parmar M, Cheruvara H, Marshall RL, Hesketh SJ, Feasey MC, Ruggerone P, Vargiu AV, Postis VLG, et al. Cryo-EM Structure and Molecular Dynamics Analysis of the Fluoroquinolone Resistant Mutant of the AcrB Transporter from Salmonella. Microorganisms. 2020; 8(6):943. https://doi.org/10.3390/microorganisms8060943
Chicago/Turabian StyleJohnson, Rachel M., Chiara Fais, Mayuriben Parmar, Harish Cheruvara, Robert L. Marshall, Sophie J. Hesketh, Matthew C. Feasey, Paolo Ruggerone, Attilio V. Vargiu, Vincent L. G. Postis, and et al. 2020. "Cryo-EM Structure and Molecular Dynamics Analysis of the Fluoroquinolone Resistant Mutant of the AcrB Transporter from Salmonella" Microorganisms 8, no. 6: 943. https://doi.org/10.3390/microorganisms8060943