Genomic and Metabolic Insights into Denitrification, Sulfur Oxidation, and Multidrug Efflux Pump Mechanisms in the Bacterium Rhodoferax sediminis sp. nov.

 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sampling, Isolation and Cultivation of the Strain
2.2. Morphological, Physiological and Chemotaxonomic Characteristics
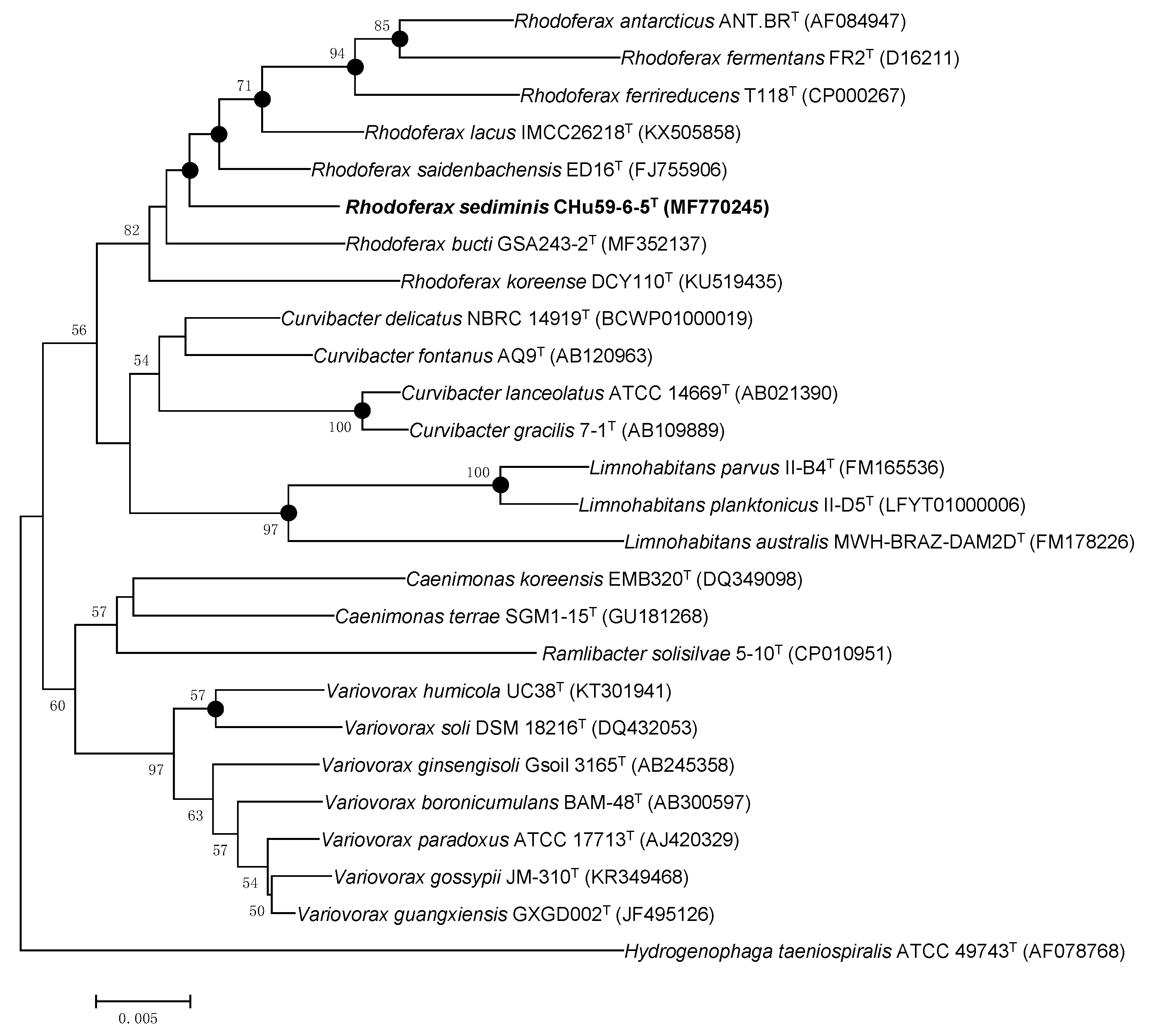
2.3. Genomic and Phylogenetic Analyses
3. Results and Discussion
3.1. Physiological Tests
3.2. Chemotaxonomy
3.3. Genomic Analysis: The Taxonomic Status
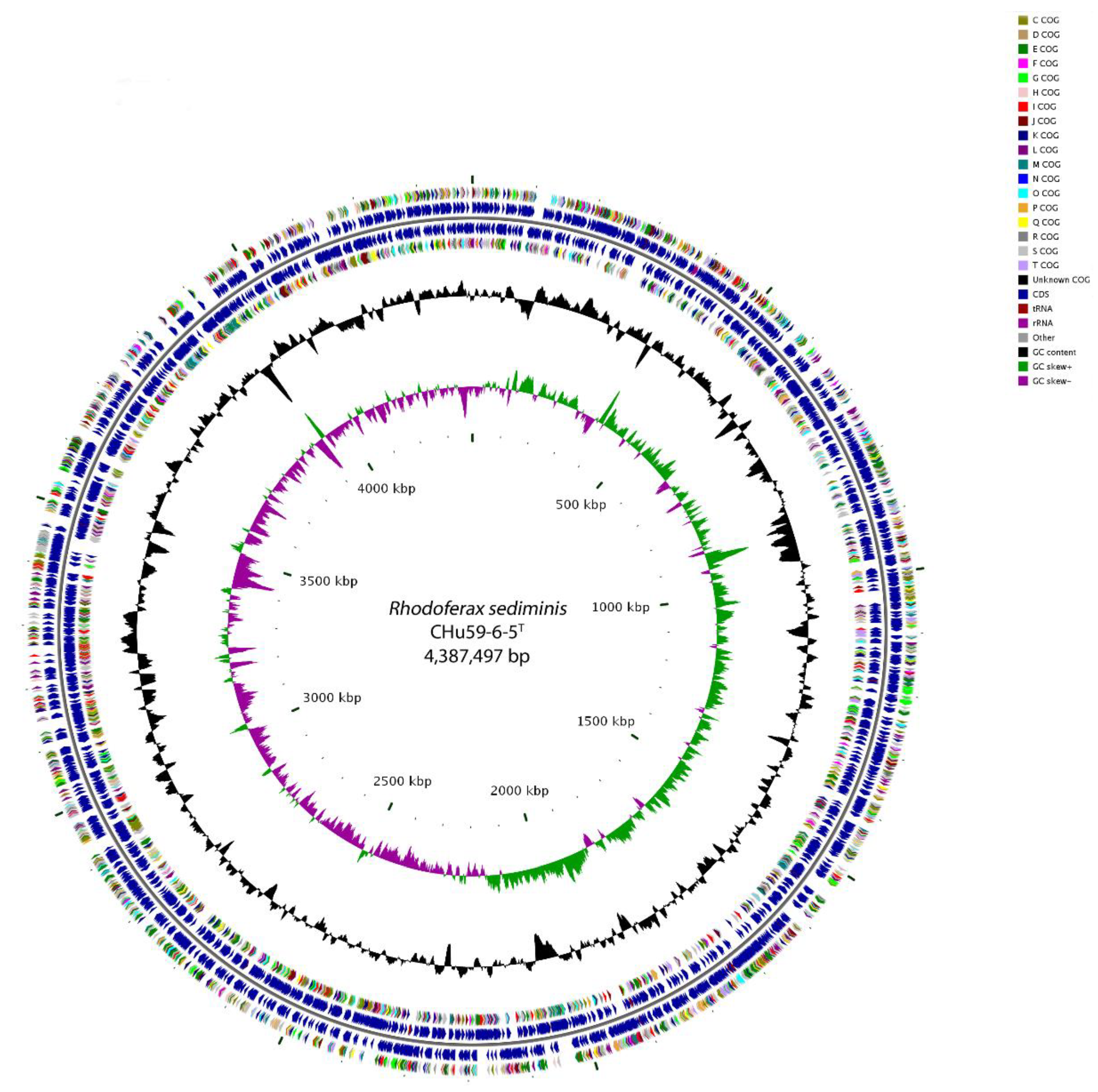
3.4. Genome Properties
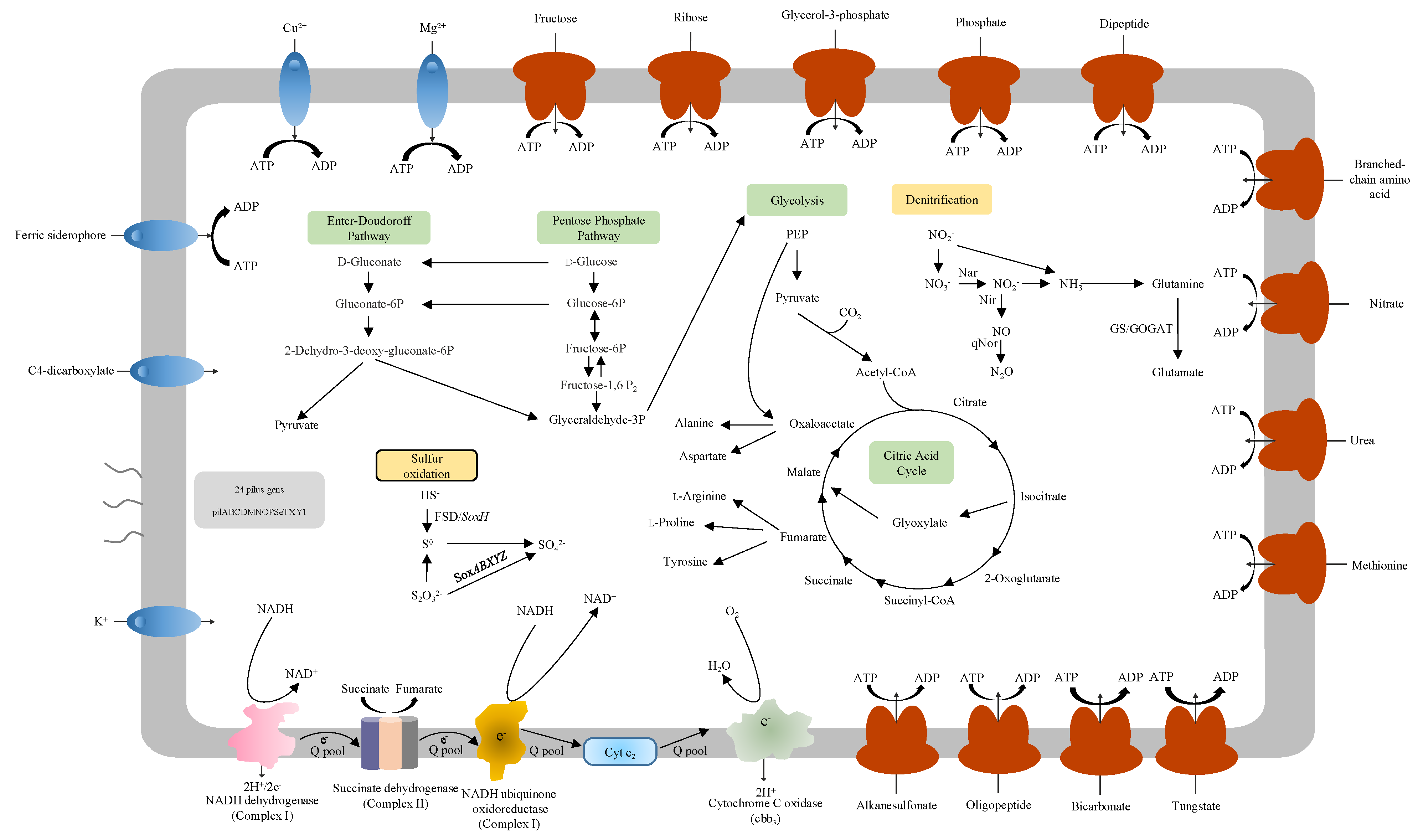
3.5. Carbon Metabolism
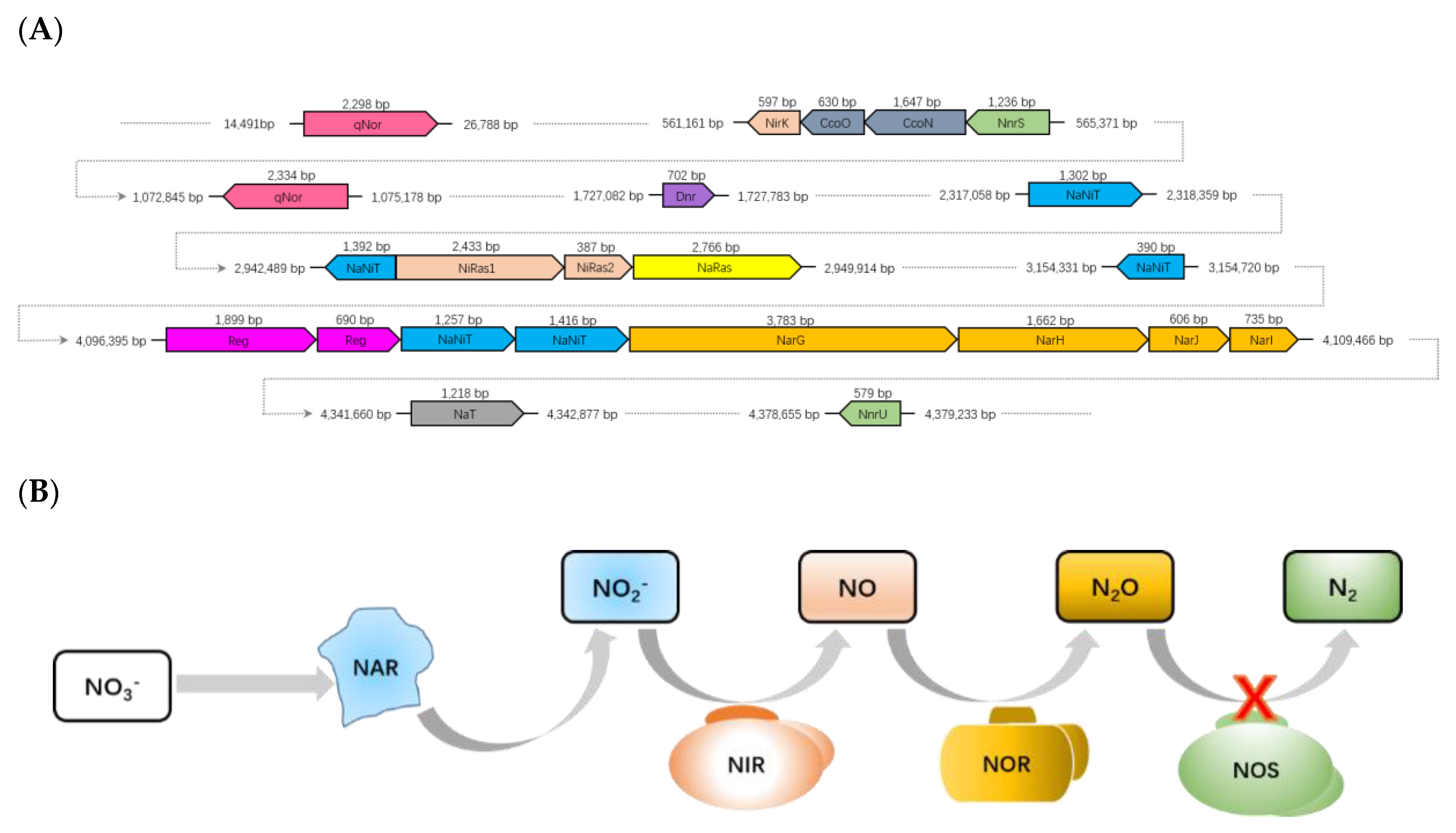
3.6. Denitrification
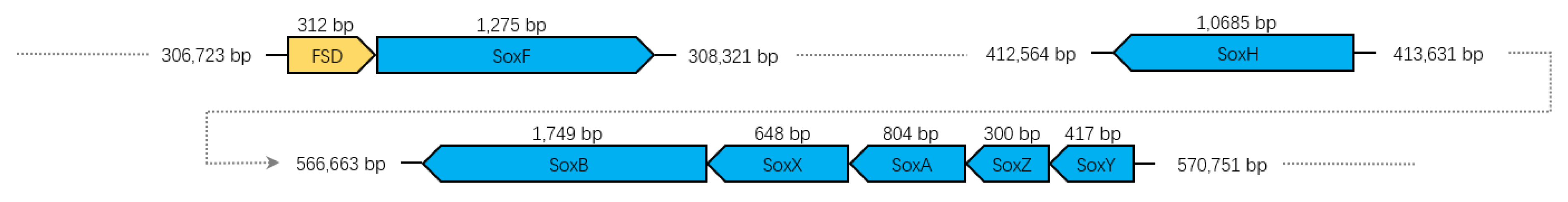
3.7. Sulfur Oxidation
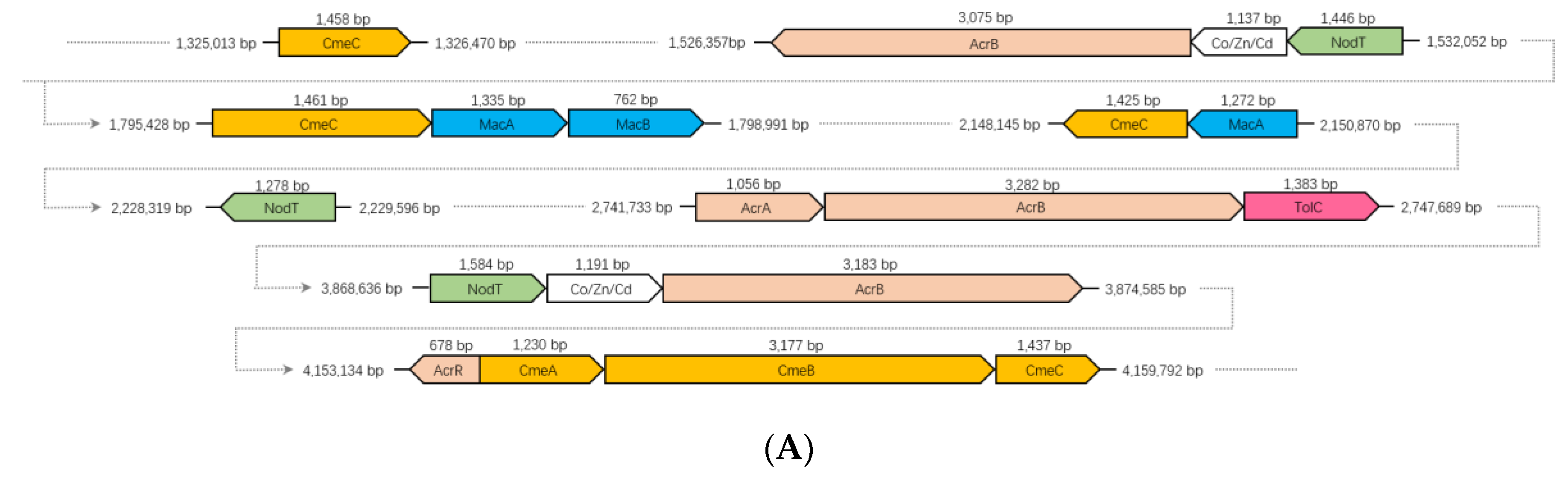
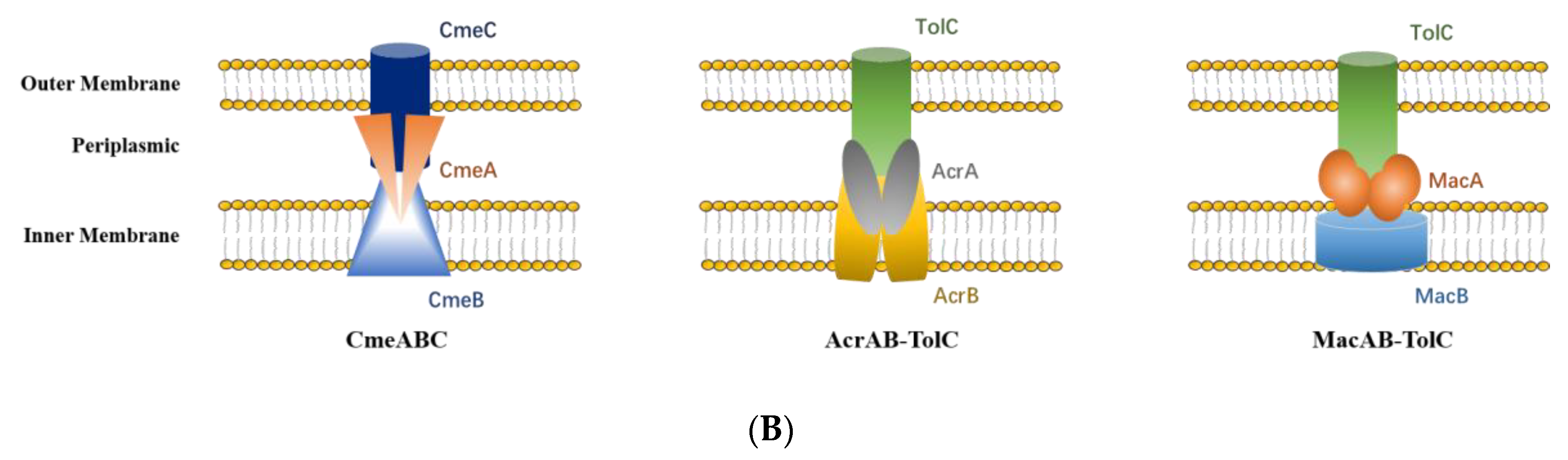
3.8. RND and ABC Efflux Systems
3.9. Motility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hiraishi, A.; Hoshino, Y.; Satoh, T. Rhodoferax fermentans gen. nov., sp. nov., a phototrophic purple nonsulfur bacterium previously referred to as the “Rhodocyclus gelatinosus-like” group. Arch. Microbiol. 1991, 155, 330–336. [Google Scholar] [CrossRef]
- Madigan, M.T.; Jung, D.O.; Woese, C.R.; Achenbach, L.A. Rhodoferax antarcticus sp. nov., a moderately psychrophilic purple nonsulfur bacterium isolated from an Antarctic microbial mat. Arch. Microbiol. 2000, 173, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Finneran, K.T.; Johnsen, C.V.; Lovley, D.R. Rhodoferax ferrireducens sp. nov., a psychrotolerant, facultatively anaerobic bacterium that oxidizes acetate with the reduction of Fe(III). Int. J. Syst. Evol. Microbiol. 2003, 53, 669–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaden, R.; Spröer, C.; Beyer, D.; Krolla-Sidenstein, P. Rhodoferax saidenbachensis sp. nov., a psychrotolerant, very slowly growing bacterium within the family Comamonadaceae, proposal of appropriate taxonomic position of Albidiferax ferrireducens strain T118T in the genus Rhodoferax and emended description of the genus Rhodoferax. Int. J. Syst. Evol. Microbiol. 2014, 64, 1186–1193. [Google Scholar]
- Farh, M.E.; Kim, Y.J.; Singh, P.; Jung, S.Y.; Kang, J.P.; Yang, D.C. Rhodoferax koreense sp. nov, an obligately aerobic bacterium within the family Comamonadaceae, and emended description of the genus Rhodoferax. J. Microbiol. 2017, 55, 767–774. [Google Scholar] [CrossRef]
- Park, M.; Song, J.; Nam, G.G.; Cho, J.C. Rhodoferax lacus sp. nov., isolated from a large freshwater lake. Int. J. Syst. Evol. Microbiol. 2019, 69, 3135–3140. [Google Scholar] [CrossRef]
- Zhou, D.; Tan, X.; Zhang, W.; Chen, H.Y.; Fan, Q.M.; He, X.L.; Lv, J. Rhodoferax bucti sp. nov., isolated from fresh water. Int. J. Syst. Evol. Microbiol. 2019, 69, 3903–3909. [Google Scholar] [CrossRef]
- Baker, J.M.; Riester, C.J.; Skinner, B.M.; Newell, A.W.; Swingley, W.D.; Madigan, M.T.; Jung, D.O.; Asao, M.; Chen, M.; Loughlin, P.C.; et al. Genome Sequence of Rhodoferax antarcticus ANT.BRT.; A psychrophilic purple nonsulfur bacterium from an Antarctic microbial mat. Microorganisms 2017, 5, 8. [Google Scholar] [CrossRef]
- Risso, C.; Sun, J.; Zhuang, K.; Mahadevan, R.; DeBoy, R.; Ismail, W.; Shrivastava, S.; Huot, H.; Kothari, S.; Daugherty, S.; et al. Genome-scale comparison and constraint-based metabolic reconstruction of the facultative anaerobic Fe(III)-reducer Rhodoferax ferrireducens. BMC Genomics 2009, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Rabaey, K.; Verstraete, W. Microbial fuel cells: Novel biotechnology for energy generation. Trends Biotechnol. 2005, 23, 291–298. [Google Scholar] [CrossRef]
- Salka, I.; Čuperová, Z.; Mašín, M.; Koblížek, M.; Grossart, H.P. Rhodoferax related pufM gene cluster dominates the aerobic anoxygenic phototrophic communities in German freshwater lakes. Environ. Microbiol. 2011, 13, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.M.; Lee, S.J.; Kim, J.H.; Kim, H.S.; Yoon, B.D. Seasonal variation and indirect monitoring of microcystin concentrations in Daechung Reservoir, Korea. Appl. Environ. Microbiol. 2001, 67, 1484–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Joung, S.H.; Ahn, C.Y.; Ko, S.R.; Boo, S.M.; Oh, H.M. Annual variation of Microcystis genotypes and their potential toxicity in water and sediment from a eutrophic reservoir. FEMS Microbiol. Ecol. 2010, 74, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, C.Y.; Oh, H.M.; Park, Y.S. Evaluation of environmental factors on cyanobacterial bloom in eutrophic reservoir using artificial neural network. J. Phycol. 2011, 47, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Lee, C.S.; Ahn, C.Y.; Lee, H.G.; Lee, S.; Shin, H.H.; Lim, D.; Oh, H.M. Abundant iron and sulfur oxidizers in the stratified sediment of a eutrophic freshwater reservoir with annual cyanobacterial blooms. Sci. Rep. 2017, 7, 43814. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Ko, S.R.; Lee, C.S.; Ahn, C.Y.; Oh, H.M.; Lee, H.G. Asprobacter aquaticus gen. nov., sp. nov., a prosthecate alphaproteobacterium isolated from fresh water. Int. J. Syst. Evol. Microbiol. 2017, 67, 4443–4448. [Google Scholar] [CrossRef]
- Lee, H.G.; Ko, S.R.; Lee, J.W.; Lee, C.S.; Ahn, C.Y.; Oh, H.M.; Jin, L. Blastomonas fulva sp. nov., aerobic photosynthetic bacteria isolated from a Microcystis culture. Int. J. Syst. Evol. Microbiol. 2017, 67, 3071–3076. [Google Scholar] [CrossRef]
- Bates, R.G.; Bower, V.E. Alkaline solutions for pH control. Anal. Chem. 1956, 28, 1322–1324. [Google Scholar] [CrossRef]
- Gomori, G. Preparation of buffers for use in enzyme studies. Methods Enzymol. 1955, 1, 138–146. [Google Scholar]
- Jin, L.; Ko, S.R.; Jin, C.Z.; Jin, F.J.; Li, T.; Ahn, C.Y.; Oh, H.M.; Lee, H.G. Description of novel members of the family Sphingomonadaceae: Aquisediminimonas profunda gen. nov., sp. nov., and Aquisediminimonas sediminicola sp. nov., isolated from freshwater sediment. Int. J. Syst. Evol. Microbiol. 2019, 69, 2179–2186. [Google Scholar] [CrossRef]
- Komagata, K.; Suzuki, K.I. Lipid and cell wall analysis in bacterial systematics. Methods Microbiol. 1987, 19, 161–207. [Google Scholar]
- Tindall, B.J. Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol. Lett. 1990, 66, 199–202. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A. Lane DJ 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBio-Cloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. mega7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method; a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limit on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Devoid, S.; Disz, T.; Edwards, R.A.; Henry, C.S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. SEED servers: High-performance access to the SEED genomes, annotations, and metabolic models. PLoS ONE 2012, 7, e48053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Waidner, L.A.; Kirchman, D.L. Aerobic anoxygenic photosynthesis genes and operons in uncultured bacteria in the Delaware River. Environ. Microbiol. 2005, 7, 1896–1908. [Google Scholar] [CrossRef]
- Tuveson, R.W.; Larson, R.A.; Kagan, J. Role of cloned carotenoid genes expressed in Escherichia coli in protecting against inactivation by near-UV light and specific phototoxic molecules. J. Bacteriol. 1988, 170, 4675–4680. [Google Scholar] [CrossRef] [Green Version]
- Badger, M.R.; Bek, E.J. Multiple Rubisco forms in proteobacteria: Their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 2008, 59, 1525–1541. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.K.; Marden, J.; Han, M.; Swingley, W.D.; Mastrian, S.D.; Chowdhury, S.R.; Hao, J.; Helmy, T.; Kim, S.; Kurdoglu, A.A.; et al. Metabolic flexibility revealed in the genome of the cyst-forming α-1 proteobacterium Rhodospirillum centenum. BMC Genom. 2010, 11, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Galloway, J.N.; Dentener, F.J.; Capone, D.G.; Boyer, E.W.; Howarth, R.W.; Seitzinger, S.P.; Asner, G.P.; Cleveland, C.C.; Green, P.A.; Holland, E.A.; et al. Nitrogen cycles: Past, present and future. Biogeochemistry 2004, 70, 153–226. [Google Scholar] [CrossRef]
- Seitzinger, S.; Harrison, J.A.; Bohlke, J.K.; Bouwman, A.F.; Lowrance, R.; Peterson, B.; Tobias, C.; Drecht, G.V. Denitrification across Landscapes and Waterscapes: A Synthesis. Ecol. Appl. 2006, 16, 2064–2090. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, W.H. On the fate of anthropogenic nitrogen. Proc. Natl. Acad. Sci. USA 2009, 106, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.; Klotz, M.G. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 2013, 1827, 114–135. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.J. Bacterial respiration: A flexible process for a changing environment. Microbiology 2000, 146, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Rinaldo, S.; Cutruzzola, F. Nitrite reductases in denitrification. Biol. Nitrogen Cycle 2007, 37–55. [Google Scholar] [CrossRef]
- Nordling, M.; Young, S.; Karlsson, B.G.; Lundberg, L.G. The structural gene for cytochrome c551 from Pseudomonas aeruginosa. The nucleotide sequence shows a location downstream of the nitrite reductase gene. FEBS Lett. 1990, 259, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Pearson, I.V.; Page, M.D.; van Spanning, R.J.; Ferguson, S.J. A mutant of Paracoccus denitrificans with disrupted genes coding for cytochrome c550 and pseudoazurin establishes these two proteins as the in vivo electron donors to cytochrome cd1 nitrite reductase. J. Bacteriol. 2003, 185, 6308–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’acqua, S.; Moura, I.; Moura, J.J.; Pauleta, S.R. The electron transfer complex between nitrous oxide reductase and its electron donors. J. Biol. Inorg. Chem. 2011, 16, 1241–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumft, W.G.; Kroneck, P.M. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by Bacteria and Archaea. Adv. Microb. Physiol. 2007, 52, 107–227. [Google Scholar]
- Sanford, R.A.; Wagner, D.D.; Wu, Q.; Chee-Sanford, J.C.; Thomas, S.H.; Cruz-García, C.; Rodríguez, G.; Massol-Deyá, A.; Krishnani, K.K.; Ritalahti, K.M. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. USA 2012, 109, 19709–19714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fike, D.A.; Bradley, A.S.; Leavitt, W.D. Geomicrobiology of sulfur. In Ehrlich’s Geomicrobiology, 6th ed.; Taylor & Francis: Boca Raton, FL, USA, 2016. [Google Scholar]
- Burgin, A.J.; Hamilton, S.K. NO3−-Driven SO42− Production in Freshwater Ecosystems: Implications for N and S Cycling. Ecosystems 2008, 11, 908–922. [Google Scholar] [CrossRef]
- Friedrich, C.G.; Bardischewsky, F.; Rother, D.; Quentmeier, A.; Fischer, J. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 2005, 8, 253–259. [Google Scholar] [CrossRef]
- Fry, B.; Ruf, W.; Gest, H.; Hayes, J.M. Sulfur isotope effects associated with oxidation of sulfide by O2 in aqueous solution. Isot. Geosci. 1988, 73, 205–210. [Google Scholar] [CrossRef]
- Wall, J.D.; Arkin, A.P.; Balci, N.C.; Rapp-Giles, B. Genetics and genomics of sulfate respiration in Desulfovibrio. Microbial. Sulfur Metabolism 2008, 1–12. [Google Scholar] [CrossRef]
- Dahl, C. Inorganic sulfur compounds as electron donors in purple sulfur bacteria. Sulfur Metab. Phototrophic Org. 2008, 27, 289–317. [Google Scholar]
- Quentmeier, A.; Hellwig, P.; Bardischewsky, F.; Grelle, G.; Kraft, R.; Friedrich, C.G. Sulfur oxidation in Paracoccus pantotrophus: Interaction of the sulfur-binding protein SoxYZ with the dimanganese SoxB protein. Biochem. Biophys. Res. Commun. 2003, 312, 1011–1018. [Google Scholar] [CrossRef]
- Epel, B.; Schäfer, K.O.; Quentmeier, A.; Friedrich, C.G.; Lubitz, W. Multifrequency EPR analysis of the dimanganese cluster of the putative sulfate thiohydrolase SoxB of Paracoccus pantotrophus. J. Biol. Inorg. Chem. 2005, 10, 636–642. [Google Scholar] [CrossRef]
- Bogomolnaya, L.M.; Andrews, K.D.; Talamantes, M.; Maple, A.; Ragoza, Y.; Vazquez-Torres, A.; Andrews-Polymenis, H. The ABC-type efflux pump MacAB protects Salmonella enterica serovar typhimurium from oxidative stress. MBio 2013, 4, e00630-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H. The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar] [PubMed]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Tipton, K.A.; Farokhyfar, M.; Rather, P.N. Multiple roles for a novel RND-type efflux system in Acinetobacter baumannii AB5075. Microbiologyopen 2016, 6, e00418. [Google Scholar] [CrossRef]
- Lin, J.; Michel, L.O.; Zhang, Q. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob. Agents Chemother. 2002, 46, 2124–2131. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Gu, R.; Su, C.C.; Routh, M.D.; Harris, K.C.; Jewell, E.S.; McDermott, G.; Yu, E.W. Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J. Mol. Biol. 2007, 374, 591–603. [Google Scholar] [CrossRef]
- Piddock, L.J. Multidrug-resistance efflux pumps—Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef]
- Lin, Y.T.; Huang, Y.W.; Liou, R.S.; Chang, Y.C.; Yang, T.C. MacABCsm, an ABC-type tripartite efflux pump of Stenotrophomonas maltophilia involved in drug resistance, oxidative and envelope stress tolerances and biofilm formation. J. Antimicrob. Chemother. 2014, 69, 3221–3226. [Google Scholar] [CrossRef]
- Greene, N.P.; Kaplan, E.; Crow, A.; Koronakis, V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front. Microbiol. 2018, 9, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turlin, E.; Heuck, G.; Simões Brandão, M.I.; Szili, N.; Mellin, J.R.; Lange, N.; Wandersman, C. Protoporphyrin (PPIX) efflux by the MacAB-TolC pump in Escherichia coli. Microbiologyopen 2014, 3, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Anes, J.; McCusker, M.P.; Fanning, S.; Martins, M. The ins and outs of RND efflux pumps in Escherichia coli. Front Microbiol. 2015, 6, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, A.J.; So, M.; Sheetz, M.P. Pilus retraction powers bacterial twitching motility. Nature 2000, 407, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Goosens, V.J.; Busch, A.; Georgiadou, M.; Castagnini, M.; Forest, K.T.; Waksman, G.; Pelicic, V. Reconstitution of a minimal machinery capable of assembling periplasmic type IV pili. Proc. Natl. Acad. Sci. USA 2017, 114, E4978–E4986. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | CHu59-6-5T | R. saidenbachensis DSM 22694T | R. lacus KACC 18983T | R. bucti KCTC 62564T | R. koreense KCTC 52288T |
|---|---|---|---|---|---|
| Isolation source | Sediment | Sediment* | Freshwater† | Freshwaterǂ | Sludge§ |
| Morphology | Short rods | Short rods* | Rods† | Curved rodsǂ | Rods§ |
| Colony colour | Colourless | Colourless | Colourless | Peach brown | Colourless |
| Motility | + | + | + | - | + |
| Growth temperature | 4–37 | 4–30* | 4–30† | 15–35ǂ | 4–30§ |
| Oxidase/catalase | +/+ | +/− | +/+ | −/+ | −/+ |
| Urease | − | + | + | − | + |
| Aesculin hydrolysis | − | − | + | + | − |
| Enzyme activity: | |||||
| alkaline phosphatase | − | + | + | + | + |
| esterase (C4) | + | + | + | + | − |
| esterase lipase (C8) | + | + | − | + | + |
| α-glucosidase | − | − | − | + | − |
| α-galactosidase | − | − | − | + | − |
| β-galactosidase | − | − | + | + | − |
| leucine arylamidase | + | − | + | + | + |
| naphthol-AS-BI-phosphohydrolase | − | + | − | + | + |
| Carbon utilization: | |||||
| l-alanine | + | − | − | − | + |
| l-arabinose | − | − | − | − | + |
| citrate | + | − | − | − | − |
| l-fucose | − | − | − | + | − |
| gluconate | − | − | − | − | + |
| d-glucose | + | + | + | − | + |
| histidine | − | − | − | − | + |
| 3-hydroxy-benzoate | − | − | − | − | + |
| 4-hydroxy-benzoate | − | − | − | − | + |
| 3-hydroxy-butyrate | + | − | − | − | + |
| d,l-lactate | + | + | − | − | + |
| 2-ketogluconate | − | − | − | + | + |
| 5-ketogluconate | − | − | − | + | + |
| maltose | − | − | − | + | − |
| d-mannitol | − | + | − | + | + |
| d-mannose | − | + | − | − | - |
| d-melibiose | − | − | − | + | - |
| l-proline | − | − | − | − | + |
| d-ribose | − | − | − | + | + |
| d-sorbitol | − | − | − | + | − |
| d-sucrose | − | − | − | + | − |
| DNA G+C content (mol%) | 64.4 | 60.3–61* | 62.3† | 61.2ǂ | 60.3§ |
| Fatty Acids | CHu59-6-5T | R. saidenbachensis DSM 22694T | R. lacus KACC 18983T | R. bucti KCTC 62564T | R. koreense KCTC 52288T |
|---|---|---|---|---|---|
| Saturated | |||||
| C11:0 | − | − | − | − | 0.9 |
| C12:0 | 3.0 | 1.3 | 1.1 | 0.5 | 12.0 |
| C13:0 | 1.3 | − | − | − | − |
| C14:0 | 2.9 | 0.2 | 0.7 | 0.3 | 1.7 |
| C15:0 | − | 0.5 | 1.3 | 1.1 | − |
| C16:0 | 21.6 | 33.6 | 28.2 | 29.3 | 24.7 |
| C17:0 | 6.9 | 0.4 | − | 1.0 | 4.6 |
| C18:0 | − | 1.2 | 1.3 | 2.0 | 0.3 |
| Unsaturated | − | ||||
| C14:1 ω5c | − | 1.3 | 0.5 | 0.9 | 3.0 |
| C15:1 ω6c | 7.5 | − | − | − | − |
| C15:1 ω6c | − | 0.2 | − | 0.5 | − |
| C16:1 ω5c | 1.8 | − | − | − | − |
| Hydroxy | − | ||||
| C8:0 3-OH | − | 1.5 | 0.8 | 0.9 | 1.8 |
| C9:0 3-OH | 0.9 | − | − | − | − |
| C10:0 3-OH | 6.4 | − | − | − | 10.7 |
| C16:1 2-OH | 1.2 | − | − | − | − |
| C17:0 3-OH | 1.2 | − | − | − | − |
| Cyclo | − | ||||
| C17:0 cyclo | 15.0 | − | − | − | 9.9 |
| Summed features‡ | − | ||||
| 3 | 29.6 | 53.6 | 59.3 | 60.2 | 26.5 |
| 8 | 0.8 | 6.1 | 4.2 | 2.8 | 2.3 |
| Attribute | CHu59-6-5T | R. saidenbachensis DSM 22694T | R. lacus KACC 18983T | R. bucti KCTC 62564T | R. koreense KCTC 52288T | R. ferrireducens T118T | R. fermentans KACC 15304T |
|---|---|---|---|---|---|---|---|
| Genome size (bp) | 4,387,497 | 4,264,855 | 4,900,405 | 3,673,501 | 5,895,641 | 4,969,784 | 4,467,741 |
| G + C content (%) | 64.4 | 60.9 | 62.3 | 61.2 | 66.2 | 59.9 | 56.9 |
| N50 | 4,387,497 | 4,264,855 | 234,657 | 937,707 | 5,800,473 | 4,712,337 | 4,447,702 |
| L50 | 1 | 1 | 6 | 2 | 1 | 1 | 1 |
| Number of contigs | 1 | 1 | 72 | 8 | 3 | 2 | 2 |
| Number of coding sequences | 4191 | 4030 | 4420 | 3436 | 5346 | 4339 | 4176 |
| Number of tRNA | 43 | 46 | 44 | 44 | 47 | 45 | 53 |
| Number of rRNA | 3 | 6 | 7 | 4 | 9 | 6 | 12 |
| ANI (%) | 100 | 75.4 | 74.9 | 74.4 | 76.9 | 75.2 | 74.3 |
| dDDH (%) | 100 | 22.1 | 21.3 | 23.3 | 20.9 | 22.3 | 22.4 |
| GenBank Accession number | CP035503 | CP019239 | QFZK00000000 | VAHD00000000 | CP019236 | CP000267 | MTJN00000000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, C.-Z.; Zhuo, Y.; Wu, X.; Ko, S.-R.; Li, T.; Jin, F.-J.; Ahn, C.-Y.; Oh, H.-M.; Lee, H.-G.; Jin, L. Genomic and Metabolic Insights into Denitrification, Sulfur Oxidation, and Multidrug Efflux Pump Mechanisms in the Bacterium Rhodoferax sediminis sp. nov. Microorganisms 2020, 8, 262. https://doi.org/10.3390/microorganisms8020262
Jin C-Z, Zhuo Y, Wu X, Ko S-R, Li T, Jin F-J, Ahn C-Y, Oh H-M, Lee H-G, Jin L. Genomic and Metabolic Insights into Denitrification, Sulfur Oxidation, and Multidrug Efflux Pump Mechanisms in the Bacterium Rhodoferax sediminis sp. nov. Microorganisms. 2020; 8(2):262. https://doi.org/10.3390/microorganisms8020262
Chicago/Turabian StyleJin, Chun-Zhi, Ye Zhuo, Xuewen Wu, So-Ra Ko, Taihua Li, Feng-Jie Jin, Chi-Yong Ahn, Hee-Mock Oh, Hyung-Gwan Lee, and Long Jin. 2020. "Genomic and Metabolic Insights into Denitrification, Sulfur Oxidation, and Multidrug Efflux Pump Mechanisms in the Bacterium Rhodoferax sediminis sp. nov." Microorganisms 8, no. 2: 262. https://doi.org/10.3390/microorganisms8020262