A Multi-Omics Protocol for Swine Feces to Elucidate Longitudinal Dynamics in Microbiome Structure and Function

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study Design
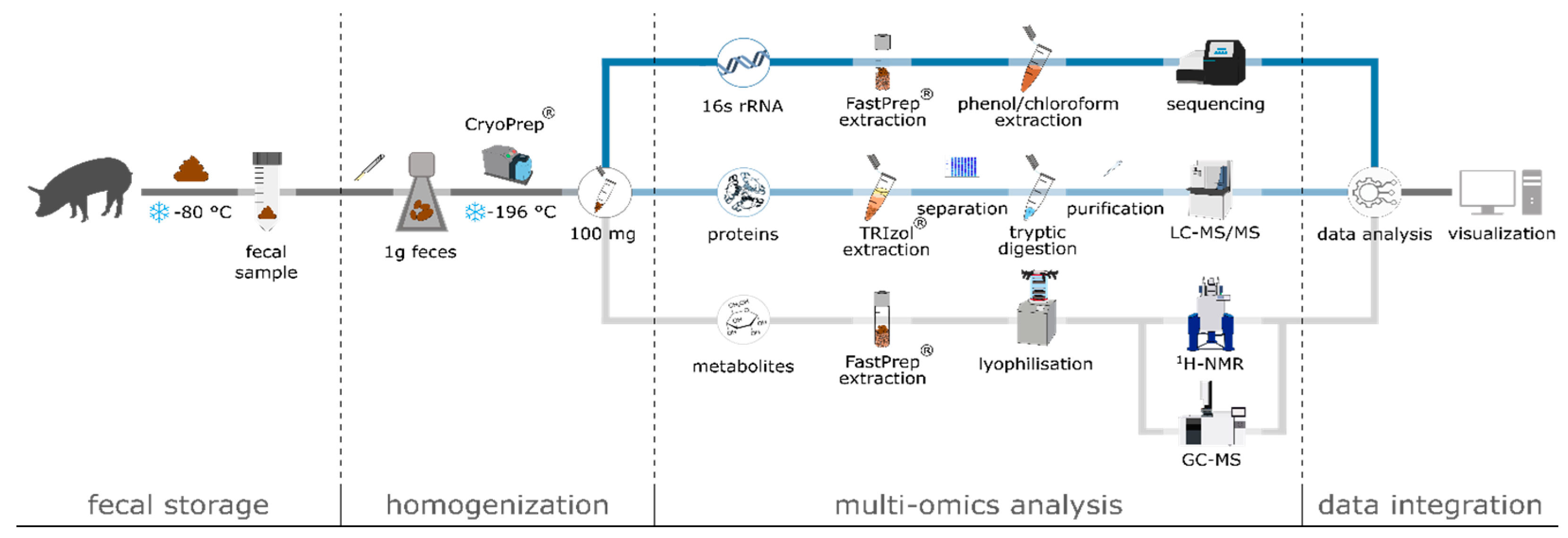
2.2. Sample Processing
2.3. DNA Extraction, 16S rRNA Gene Amplicon Library Preparation, Sequencing, and Bioinformatic Processing
2.4. Protein Extraction and Preparation for Mass Spectrometry
2.5. Mass Spectrometry Analysis
2.6. Database Assembly and Data Analysis
2.7. Taxonomic, Functional and Statistical Analysis of Protein Groups
2.8. Extraction of Metabolites
2.9. Analysis of Metabolites Using GC-MS and 1H-NMR
2.10. Swine Feces Processing Pipeline Suitable for Multi-Omics Analyses
3. Results and Discussion
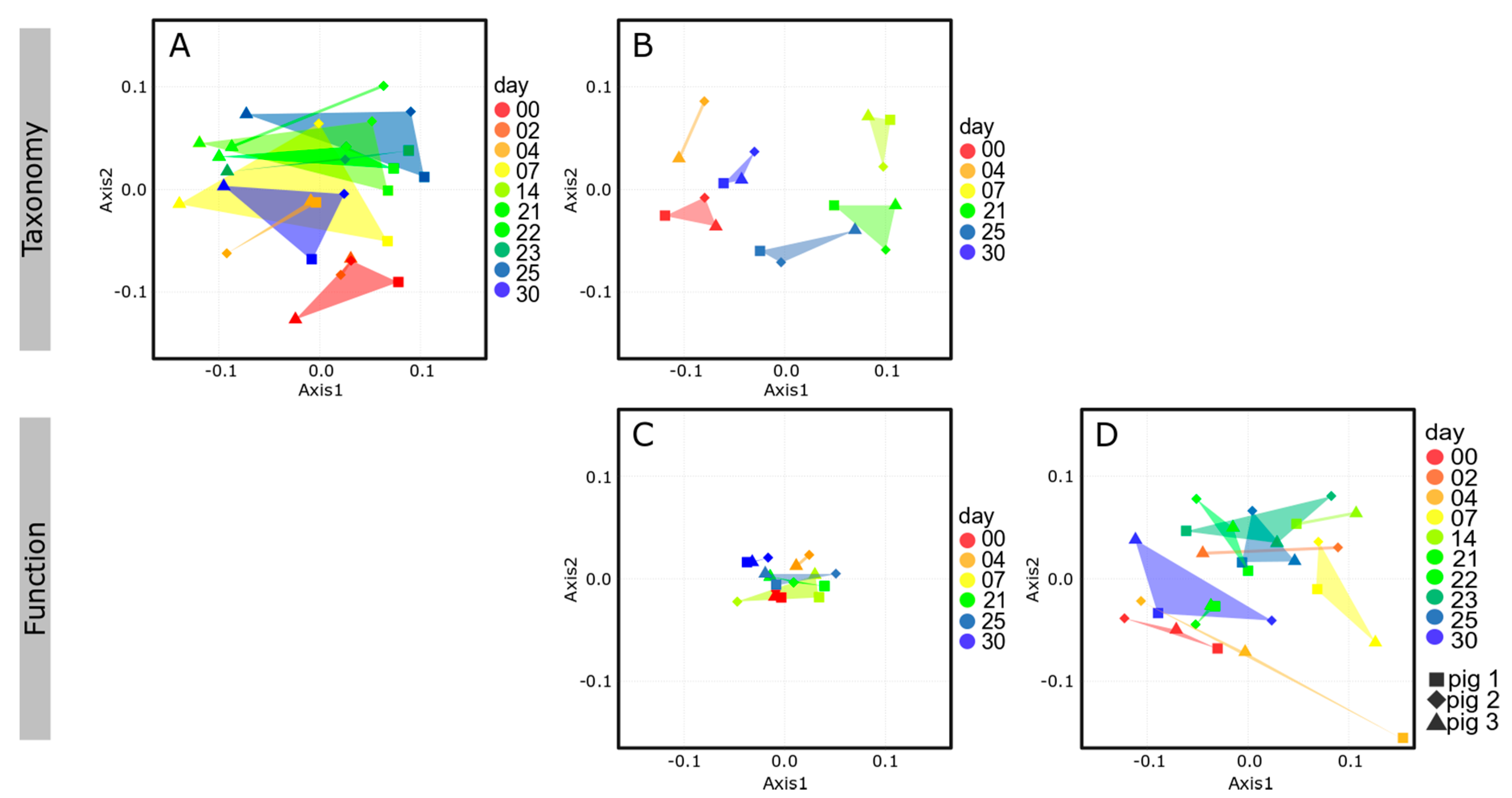
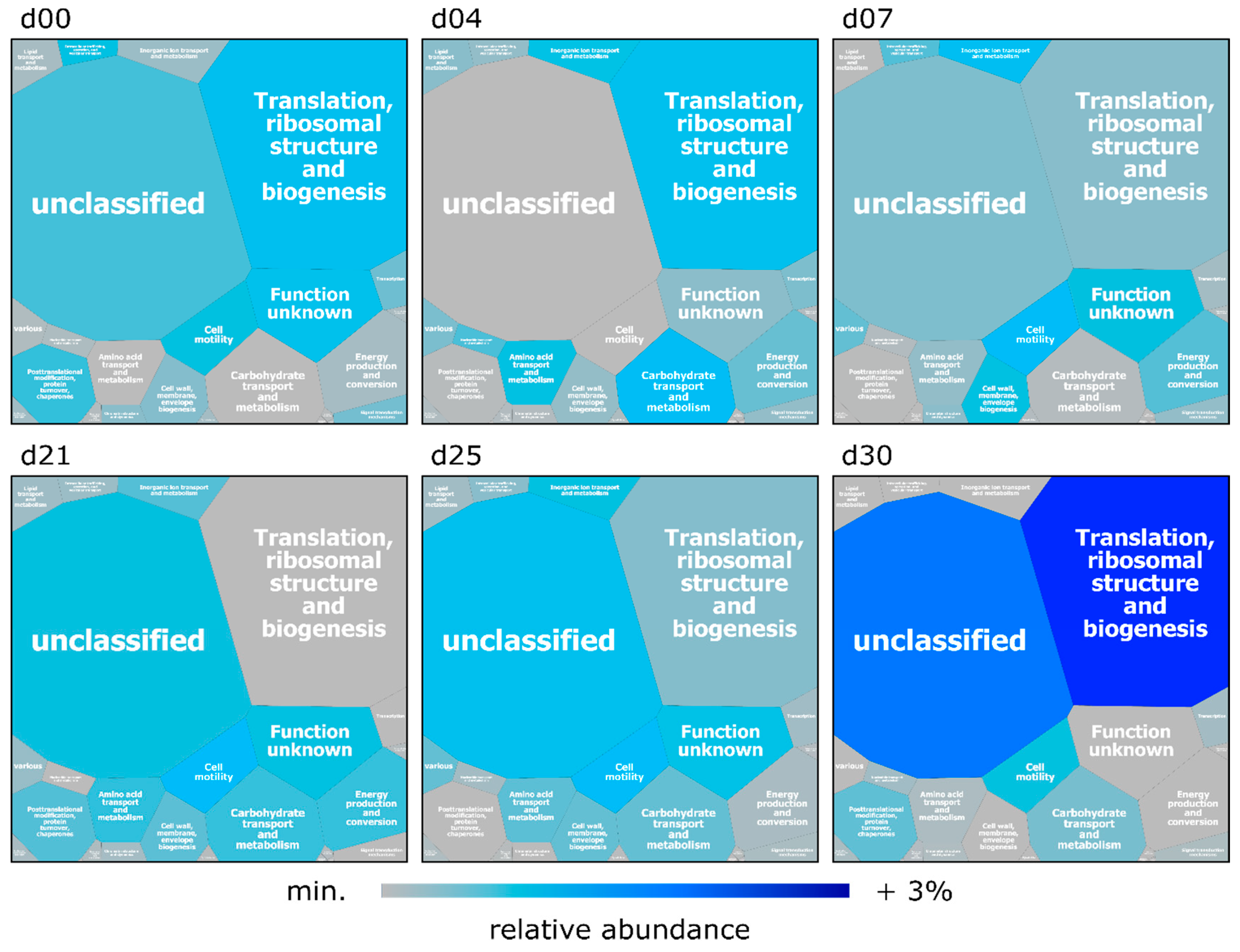
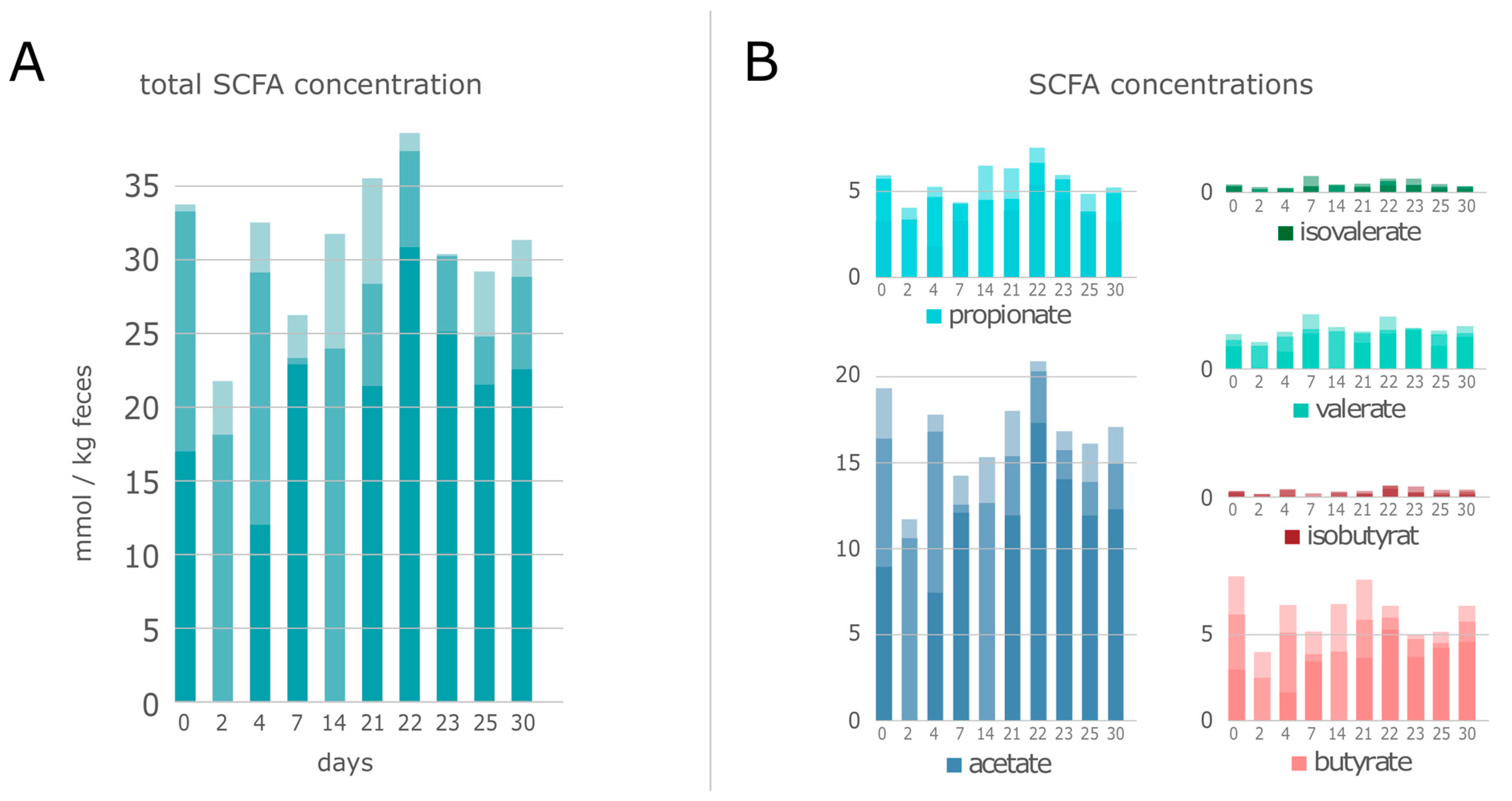
3.1. Multi-Omics Analyses of Swine Feces
3.2. Taxonomic Composition of the Intestinal Microbiome
High Similarity of the Fecal and Colonal Microbiota
3.3. Functional Analysis of the Microbiome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system: Envisioning the future. Nat. Cell Biol. 2011, 474, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V. Commensal Host-Bacterial Relationships in the Gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- DiBaise, J.K.; Frank, D.N.; Mathur, R. Impact of the Gut Microbiota on the Development of Obesity: Current Concepts. Am. J. Gastroenterol. Suppl. 2012, 1, 22–27. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Tlaskalová-Hogenová, H.; Stěpánková, R.; Kozáková, H.; Hudcovic, T.; Vannucci, L.; Tučková, L.; Rossmann, P.; Hrnčíř, T.; Kverka, M.; Zákostelská, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell. Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nat. Cell Biol. 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Sartor, R.B. Microbial Influences in Inflammatory Bowel Diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Littman, D.R.; Pamer, E.G. Role of the Commensal Microbiota in Normal and Pathogenic Host Immune Responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef] [Green Version]
- De Rodas, B.; Youmans, B.P.; Danzeisen, J.L.; Tran, H.; Johnson, T.J. Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 2018, 96, 1778–1794. [Google Scholar] [CrossRef]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Louis, S.; Rings, A.; Messner, S.; Camarinha-Silva, A.; Seifert, J.; Bischoff, S.C.; et al. Intestinal Microbiota and Microbial Metabolites Are Changed in a Pig Model Fed a High-Fat/Low-Fiber or a Low-Fat/High-Fiber Diet. PLoS ONE 2016, 11, e0154329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, M.; Dhillon, P. Using the mouse to model human disease: Increasing validity and reproducibility. Dis. Model. Mech. 2016, 9, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Niemann, H.; Petersen, B. The production of multi-transgenic pigs: Update and perspectives for xenotransplantation. Transgenic Res. 2016, 25, 361–374. [Google Scholar] [CrossRef]
- Wang, M.; Donovan, S. Human Microbiota-Associated Swine: Current Progress and Future Opportunities. ILAR J. 2015, 56, 63–73. [Google Scholar] [CrossRef]
- Isaacson, R.E.; Kim, H.B. The intestinal microbiome of the pig. Anim. Heal. Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis To Define a Core Microbiota in the Swine Gut. mSystems 2017, 2, e00004-17. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lang, T.; Shen, J.; Dai, J.; Tian, L.; Wang, X. Core Gut Bacteria Analysis of Healthy Mice. Front. Microbiol. 2019, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Roume, H.; Muller, E.E.L.; Cordes, T.; Renaut, J.; Hiller, K.; Wilmes, P. A biomolecular isolation framework for eco-systems biology. ISME J. 2013, 7, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; De Beaufort, C.; et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2017, 2, 16180. [Google Scholar] [CrossRef]
- Deusch, S.; Seifert, J. Catching the tip of the iceberg—Evaluation of sample preparation protocols for metaproteomic studies of the rumen microbiota. Proteomics 2015, 15, 3590–3595. [Google Scholar] [CrossRef]
- Tröscher-Mußotter, J.; Tilocca, B.; Stefanski, V.; Seifert, J. Analysis of the Bacterial and Host Proteins along and across the Porcine Gastrointestinal Tract. Proteomes 2019, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-N.; Pu, J.-C.; Liu, L.-X.; Wang, G.-W.; Zhou, X.-Y.; Zhang, Y.-Q.; Liu, Y.-Y.; Xie, P. Integrated Metabolomics and Proteomics Analysis Revealed Second Messenger System Disturbance in Hippocampus of Chronic Social Defeat Stress Rat. Front. Neurosci. 2019, 13, 247. [Google Scholar] [CrossRef]
- Kan, J.; Hanson, T.E.; Ginter, J.M.; Wang, K.; Chen, F. Metaproteomic analysis of Chesapeake Bay microbial communities. Saline Syst. 2005, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [Green Version]
- Kolmeder, C.; De Been, M.; Nikkilä, J.; Ritamo, I.; Mättö, J.; Valmu, L.; Salojärvi, J.; Palva, A.; Salonen, A.; De Vos, W.M. Comparative Metaproteomics and Diversity Analysis of Human Intestinal Microbiota Testifies for Its Temporal Stability and Expression of Core Functions. PLoS ONE 2012, 7, e29913. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, T.; Sehl, J.; Karte, C.; Schäfer, A.; Hühr, J.; Mettenleiter, T.C.; Schröder, C.; Köllner, B.; Ulrich, R.; Blohm, U. Experimental H1N1pdm09 infection in pigs mimics human seasonal influenza infections. PLoS ONE 2019, 14, e0222943. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Schwab, C.; Milinovich, G.; Reichert, J.; Ben Mahfoudh, K.; Decker, T.; Engel, M.; Hai, B.; Hainzl, E.; Heider, S.; et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012, 6, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2015, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: http://www.R-project.org/ (accessed on 7 January 2020).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; A Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–537. [Google Scholar]
- Smith, P.; Krohn, R.; Hermanson, G.; Mallia, A.; Gartner, F.; Provenzano, M.; Fujimoto, E.; Goeke, N.; Olson, B.; Klenk, D. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Wiechelman, K.J.; Braun, R.D.; Fitzpatrick, J.D. Investigation of the bicinchoninic acid protein assay: Identification of the groups responsible for color formation. Anal. Biochem. 1988, 175, 231–237. [Google Scholar] [CrossRef]
- Kessler, R.J.; Fanestil, D.D. Interference by lipids in the determination of protein using bicinchoninic acid. Anal. Biochem. 1986, 159, 138–142. [Google Scholar] [CrossRef]
- Brown, R.E.; Jarvis, K.L.; Hyland, K.J. Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal. Biochem. 1989, 180, 136–139. [Google Scholar] [CrossRef]
- Neuhoff, V.; Arold, N.; Taube, D.; Ehrhardt, W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 1988, 9, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lassek, C.; Burghartz, M.; Chaves-Moreno, D.; Otto, A.; Hentschker, C.; Fuchs, S.; Bernhardt, J.; Jauregui, R.; Neubauer, R.; Becher, D.; et al. A Metaproteomics Approach to Elucidate Host and Pathogen Protein Expression during Catheter-Associated Urinary Tract Infections (CAUTIs). Mol. Cell. Proteom. 2015, 14, 989–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.; Holton, K.; Nair, S.; Schlauch, D.; Sinha, R.; Quackenbush, J. MeV: MultiExperiment Viewer. In Biomedical Informatics for Cancer Research; Springer: Berlin/Heidelberg, Germany, 2010; pp. 267–277. [Google Scholar]
- Schultz, D.; Schlüter, R.; Gerth, U.; Lalk, M. Metabolic Perturbations in a Bacillus subtilis clpP Mutant during Glucose Starvation. Metabolites 2017, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Dörries, K.; Schlueter, R.; Lalk, M. Impact of Antibiotics with Various Target Sites on the Metabolome of Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 7151–7163. [Google Scholar] [CrossRef] [Green Version]
- Deda, O.; Chatziioannou, A.C.; Fasoula, S.; Palachanis, D.; Raikos, Ν.; Theodoridis, G.A.; Gika, H.G. Sample preparation optimization in fecal metabolic profiling. J. Chromatogr. B 2017, 1047, 115–123. [Google Scholar] [CrossRef]
- Matysik, S.; Le Roy, C.I.; Liebisch, G.; Claus, S.P. Metabolomics of fecal samples: A practical consideration. Trends Food Sci. Technol. 2016, 57, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, K.S.; Maier, T.V.; Walker, A.; Heinzmann, S.S.; Forcisi, S.; Martinez, I.; Walter, J.; Schmitt-Kopplin, P. Challenges of metabolomics in human gut microbiota research. Int. J. Med Microbiol. 2016, 306, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Wylezich, C.; Papa, A.; Beer, M.; Höper, D. A Versatile Sample Processing Workflow for Metagenomic Pathogen Detection. Sci. Rep. 2018, 8, 13108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.; Benndorf, D.; Rapp, E.; Reichl, U.; Palese, L.; Pollice, A. Metaproteome analysis of sewage sludge from membrane bioreactors. Proteomics 2011, 11, 2738–2744. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Pisanu, S.; Deligios, M.; Fraumene, C.; Manghina, V.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. A straightforward and efficient analytical pipeline for metaproteome characterization. Microbiome 2014, 2, 49. [Google Scholar] [CrossRef]
- Wu, J.; An, Y.; Yao, J.; Wang, Y.; Tang, H. An optimised sample preparation method for NMR-based faecal metabonomic analysis. Analyst 2010, 135, 1023–1030. [Google Scholar] [CrossRef]
- Gao, X.; Pujos-Guillot, E.; Martin, J.-F.; Galan, P.; Juste, C.; Jia, W.; Sebedio, J.-L. Metabolite analysis of human fecal water by gas chromatography/mass spectrometry with ethyl chloroformate derivatization. Anal. Biochem. 2009, 393, 163–175. [Google Scholar] [CrossRef]
- Tan, S.C.; Chong, C.W.; Yap, I.K.S.; Thong, K.L.; Teh, C.S.J. Comparative assessment of faecal microbial composition and metabonome of swine, farmers and human control. Sci. Rep. 2020, 10, 8997. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, H.; Wang, S.; Zhang, W.; Wang, J.; Tian, H.; Wang, Y.; Ji, H. Fecal Microbiota and Its Correlation With Fatty Acids and Free Amino Acids Metabolism in Piglets After a Lactobacillus Strain Oral Administration. Front. Microbiol. 2019, 10, 785. [Google Scholar] [CrossRef]
- Wen, X.; Wang, L.; Zheng, C.; Yang, X.; Ma, X.; Wu, Y.; Chen, Z.; Jiang, Z. Fecal scores and microbial metabolites in weaned piglets fed different protein sources and levels. Anim. Nutr. 2018, 4, 31–36. [Google Scholar] [CrossRef]
- Leser, T.D.; Amenuvor, J.Z.; Jensen, T.K.; Lindecrona, R.H.; Boye, M.; Møller, K. Culture-Independent Analysis of Gut Bacteria: The Pig Gastrointestinal Tract Microbiota Revisited. Appl. Environ. Microbiol. 2002, 68, 673–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, E.; Aumiller, T.; Spindler, H.K.; Rosenfelder, P.; Eklund, M.; Witzig, M.; Jørgensen, H.; Knudsen, K.E.B.; Mosenthin, R. Wheat and barley differently affect porcine intestinal microbiota. J. Sci. Food Agric. 2015, 96, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Le Sciellour, M.; Renaudeau, D.; Zemb, O. Longitudinal Analysis of the Microbiota Composition and Enterotypes of Pigs from Post-Weaning to Finishing. Microorganisms 2019, 7, 622. [Google Scholar] [CrossRef] [Green Version]
- Heinritz, S.N.; Mosenthin, R.; Weiss, E. Use of pigs as a potential model for research into dietary modulation of the human gut microbiota. Nutr. Res. Rev. 2013, 26, 191–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; O Bayles, D.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, S.; Tilocca, B.; Camarinha-Silva, A.; Seifert, J. News in livestock research—Use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Comput. Struct. Biotechnol. J. 2015, 13, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.; Szklarczyk, D.; Trachana, K.; Roth, A.; Kuhn, M.; Muller, J.; Arnold, R.; Rattei, T.; Letunic, I.; Doerks, T.; et al. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012, 40, D284–D289. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.R.; Heinken, A.; Scott, K.P.; Swann, J.; Thiele, I.; Tuohy, K.M. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Allison, C.; Gibson, S.A.W.; Cummings, J.H. Contribution of the microflora to proteolysis in the human large intestine. J. Appl. Bacteriol. 1988, 64, 37–46. [Google Scholar] [CrossRef]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Liu, L.; Guo, X.; Rao, J.N.; Zou, T.; Xiao, L.; Yu, T.; Timmons, J.A.; Turner, D.J.; Wang, J.-Y. Polyamines regulate E-cadherin transcription through c-Myc modulating intestinal epithelial barrier function. Am. J. Physiol. Physiol. 2009, 296, C801–C810. [Google Scholar] [CrossRef] [Green Version]
- Löser, C.; Eisel, A.; Harms, D.; Fölsch, U.R. Dietary polyamines are essential luminal growth factors for small intestinal and colonic mucosal growth and development. Gut 1999, 44, 12–16. [Google Scholar] [CrossRef]
- Besten, G.D.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Gil Quintero, A.; Siegel, D.; Permentier, H.; Reijngoud, D.-J.; Dekker, F.W.; Bischoff, R. Stability of energy metabolites-An often overlooked issue in metabolomics studies: A review. Electrophoresis 2015, 36, 2156–2169. [Google Scholar] [CrossRef]
- Darzi, Y.; Letunic, I.; Bork, P.; Yamada, T. iPath3.0: Interactive pathways explorer v3. Nucleic Acids Res. 2018, 46, W510–W513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parada Venegas, D.; de La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canibe, N.; Steien, S.H.; Overland, M.; Jensen, B.B. Effect of K-diformate in starter diets on acidity, microbiota, and the amount of organic acids in the digestive tract of piglets, and on gastric alterations. J. Anim. Sci. 2001, 79, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.-S.R.; de Jonge, N.; Nielsen, J.L.; Højberg, O.; Lauridsen, C.; Cutting, S.M.; Canibe, N. Impact of Bacillus spp. spores and gentamicin on the gastrointestinal microbiota of suckling and newly weaned piglets. PLoS ONE 2018, 13, e0207382. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Ordaz, A.A.; Molist, F.; Hermes, R.G.; Gómez de Segura, A.; La Ragione, R.M.; Woodward, M.J.; Tchorzewska, M.A.; Collins, J.W.; Pérez, J.F.; Martín-Orúe, S.M. Effect of inclusion of lactulose and Lactobacillus plantarum on the intestinal environment and performance of piglets at weaning. Anim. Feed. Sci. Technol. 2013, 185, 160–168. [Google Scholar] [CrossRef]
- Molist, F.; de Segura, A.G.; Gasa, J.; Hermes, R.G.; Manzanilla, E.G.; Anguita, M.; Pérez, J.F. Effects of the insoluble and soluble dietary fibre on the physicochemical properties of digesta and the microbial activity in early weaned piglets. Anim. Feed. Sci. Technol. 2009, 149, 346–353. [Google Scholar] [CrossRef]
- Sakata, T. Pitfalls in short-chain fatty acid research: A methodological review. Anim. Sci. J. 2019, 90, 3–13. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Days after Starting Point | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 * | 4 ** | 7 | 14 * | 21 | 22 | 23 | 25 | 30 | ||
| Fecal samples (n = 3) | 3 | 2 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | |
| Individual number of analyzed samples after homogenization | 16S rRNA gene analysis | 3 | 2 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 |
| Metaproteomics | 3 | - | 2 | 3 | - | 3 | - | - | 3 | 3 | |
| Metabolomics | 3 | 2 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | |
| Days after Sampling | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 * | 7 | 14 * | 21 | 22 | 23 | 25 | 30 | |
| No. of ASVs ± sd; CV | 831 ± 97; 0.12 | 716 ± 112; 0.16 | 773 ± 36; 0.05 | 816 ± 169; 0.21 | 859 ± 161; 0.19 | 841 ± 88; 0.1 | 828 ± 80; 0.1 | 857 ± 161; 0.19 | 901 ± 99; 0.11 | 780 ± 47; 0.06 |
| Shannon ± sd; CV | 5.19 ± 0.10; 0.02 | 5.00 ± 0.12; 0.02 | 5.10 ± 0.13; 0.02 | 5.30 ± 0.26; 0.05 | 5.26 ± 0.23; 0.04 | 5.14 ± 0.17; 0.03 | 5.13 ± 0.13; 0.03 | 5.23 ± 0.13; 0.03 | 5.40 ± 0.14; 0.03 | 5.02 ± 0.05; 0.01 |
| Simpson ± sd; CV | 0.99 ± 0.001; 0.001 | 0.98 ± 0.003; 0.004 | 0.99 ± 0.002; 0.002 | 0.99 ± 0.003; 0.003 | 0.99 ± 0.003; 0.003 | 0.98 ± 0.003; 0.03 | 0.98 ± 0.003; 0.003 | 0.98 ± 0.003; 0.003 | 0.99 ± 0.001; 0.001 | 0.98 ± 0.002; 0.002 |
| Evenness ± sd; CV | 0.77 ± 0.006; 0.007 | 0.76 ± 0.001; 0.001 | 0.77 ± 0.021; 0.027 | 0.79 ± 0.015; 0.019 | 0.78 ± 0.013; 0.016 | 0.76 ± 0.013; 0.017 | 0.76 ± 0.009; 0.012 | 0.78 ± 0.016; 0.021 | 0.79 ± 0.011; 0.013 | 0.75 ± 0.012; 0.016 |
| Days after Sampling | ||||||
|---|---|---|---|---|---|---|
| 0 | 4 * | 7 | 21 | 25 | 30 | |
| Pig 1 | 5228 | 6215 | 5038 | 4224 | 4886 | |
| unique | 1091 | 2653 | 1279 | 960 | 1108 | |
| Pig 2 | 5342 | 6920 | 3672 | 5274 | 5450 | 6101 |
| unique | 1231 | 3303 | 1059 | 1458 | 2068 | 2217 |
| Pig 3 | 6271 | 5686 | 4837 | 5022 | 5204 | 5298 |
| unique | 2052 | 2069 | 1539 | 1601 | 1842 | 1289 |
| In 2 of 3 | 4797 | 3617 | 3882 | 4380 | 3970 | 4491 |
| Pathway | Number of Detected Metabolites |
|---|---|
| carbon core metabolism | 13 |
| amino acids | 18 |
| amino acid degradation | 6 |
| fatty acids | 6 |
| short-chain fatty acids | 6 |
| various | 21 |
| ∑ | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gierse, L.C.; Meene, A.; Schultz, D.; Schwaiger, T.; Karte, C.; Schröder, C.; Wang, H.; Wünsche, C.; Methling, K.; Kreikemeyer, B.; et al. A Multi-Omics Protocol for Swine Feces to Elucidate Longitudinal Dynamics in Microbiome Structure and Function. Microorganisms 2020, 8, 1887. https://doi.org/10.3390/microorganisms8121887
Gierse LC, Meene A, Schultz D, Schwaiger T, Karte C, Schröder C, Wang H, Wünsche C, Methling K, Kreikemeyer B, et al. A Multi-Omics Protocol for Swine Feces to Elucidate Longitudinal Dynamics in Microbiome Structure and Function. Microorganisms. 2020; 8(12):1887. https://doi.org/10.3390/microorganisms8121887
Chicago/Turabian StyleGierse, Laurin Christopher, Alexander Meene, Daniel Schultz, Theresa Schwaiger, Claudia Karte, Charlotte Schröder, Haitao Wang, Christine Wünsche, Karen Methling, Bernd Kreikemeyer, and et al. 2020. "A Multi-Omics Protocol for Swine Feces to Elucidate Longitudinal Dynamics in Microbiome Structure and Function" Microorganisms 8, no. 12: 1887. https://doi.org/10.3390/microorganisms8121887