Genomic Insights into the Fungal Lignocellulolytic Machinery of Flammulina rossica


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain, Culture, and Genomic DNA Isolation
2.2. Genome Sequencing, De Novo Assembly, Gene Prediction, and Annotation
2.3. Ortholog Analysis
2.4. CAZyme and Signal Peptide Identification
2.5. Data Access
3. Results and Discussion
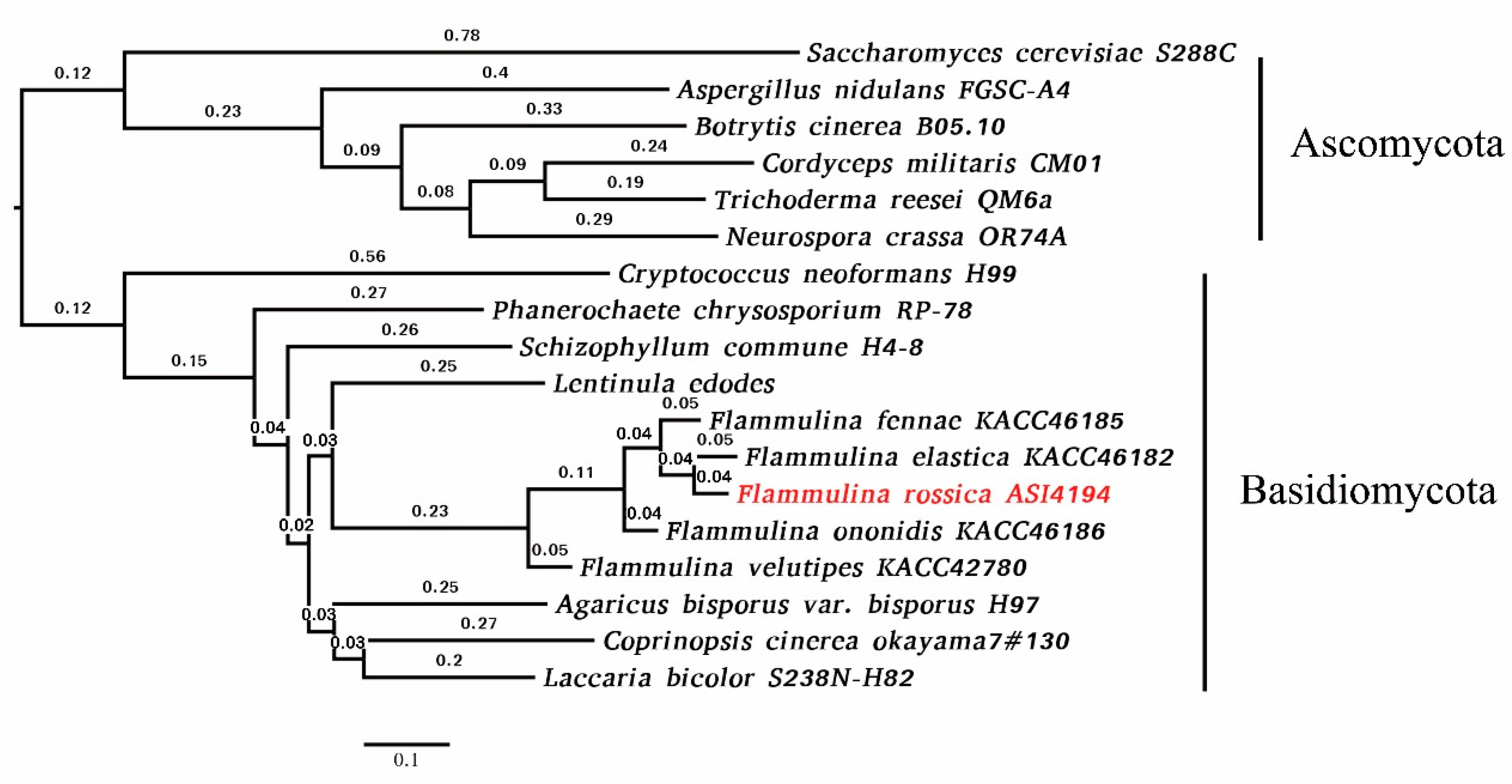
3.1. De Novo Genome Assembly, Gene Prediction, and Genome Comparisons
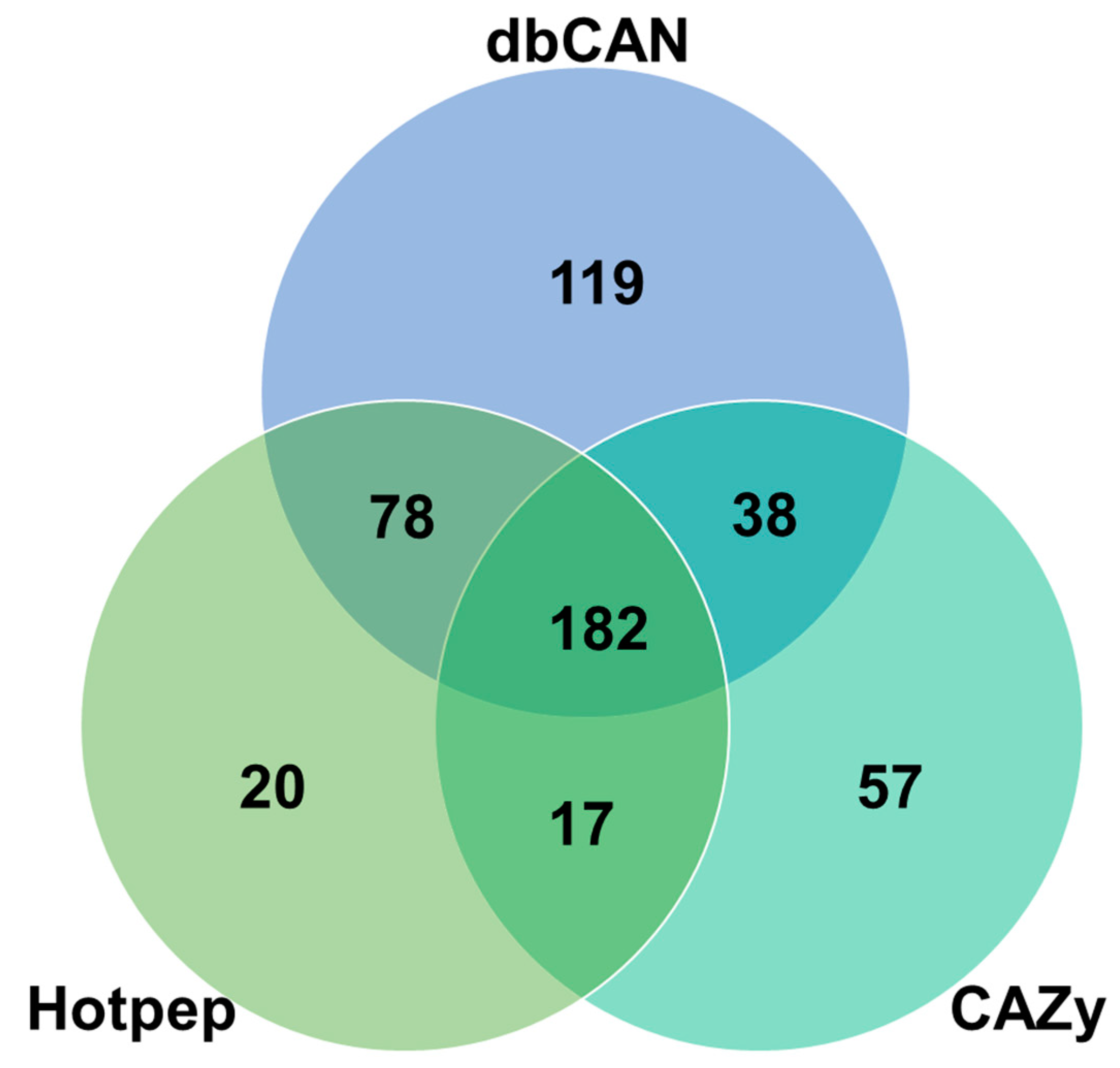
3.2. F. rossica CAZymes and Genome-Wide Comparisons with Other Fungal Species
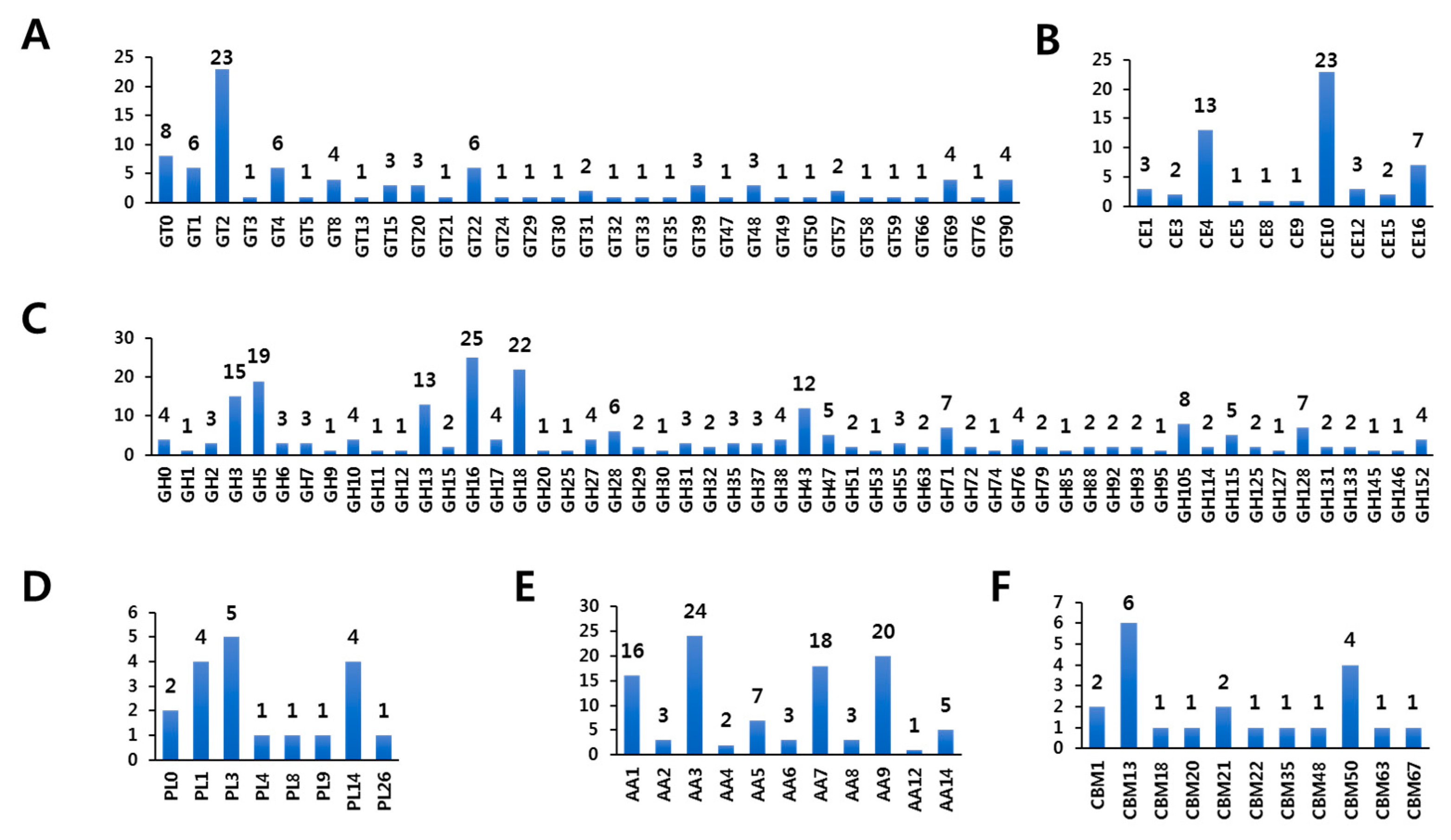
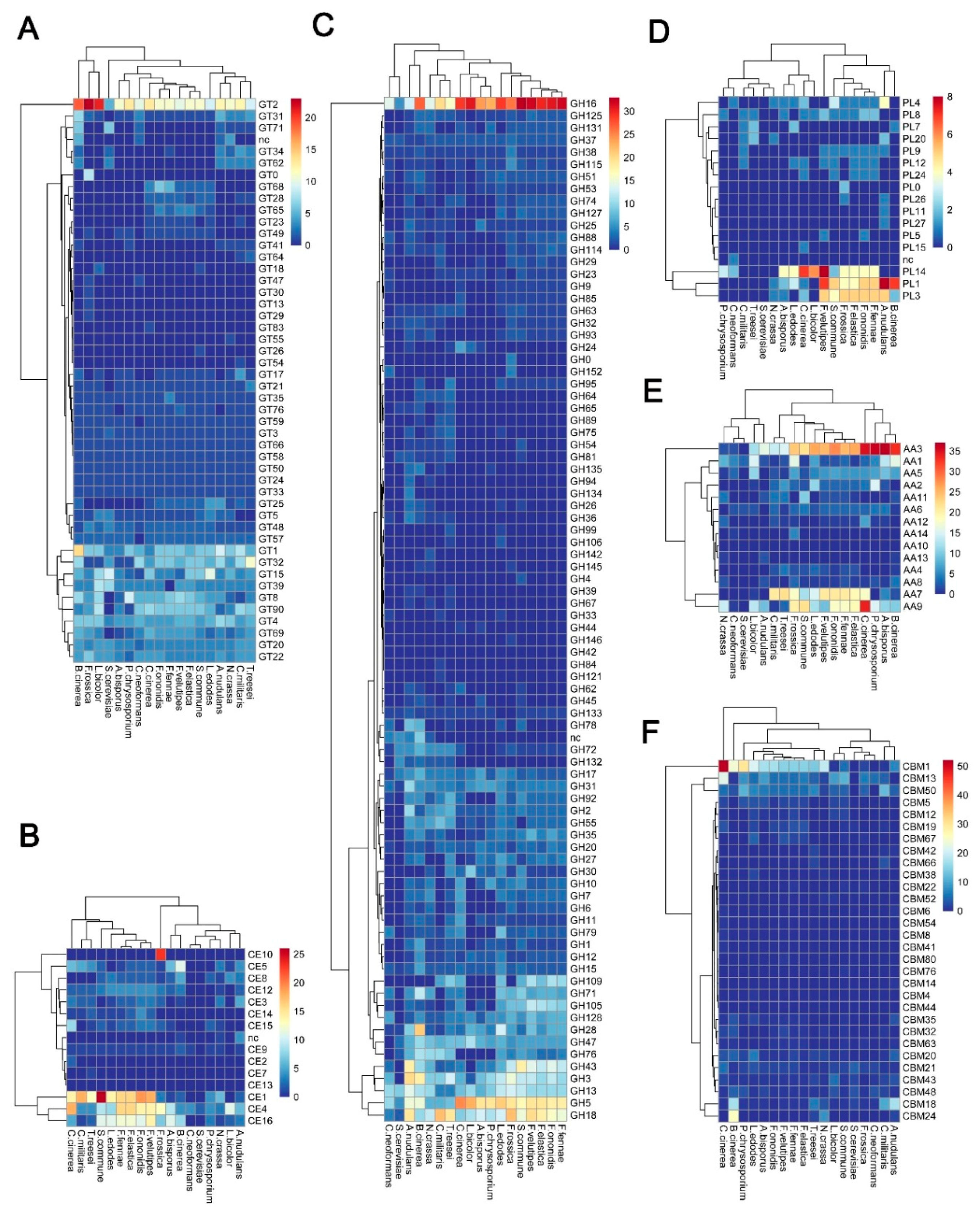
3.2.1. Glycosyltransferases (GTs) of F. rossica Genome
3.2.2. Carbohydrate Esterases (CEs) of the F. rossica Genome
3.2.3. Glycoside Hydrolases (GHs) of F. rossica Genome
3.2.4. Polysaccharide Lyases (PLs) of the F. rossica Genome
3.2.5. Auxiliary Activities (AAs) of F. rossica Genome
3.2.6. Carbohydrate-Binding Modules (CBMs) of F. rossica Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Redhead, S.A.; Petersen, R.H. New species, varieties and combinations in the genus Flammulina. Mycotaxon 1999, 71, 285–294. [Google Scholar]
- Hughes, K.W.; McGhee, L.L.; Methven, A.S.; Johnson, J.E.; Petersen, R.H. Patterns of geographic speciation in the genus Flammulina based on sequences of the ribosomal ITS-5.8S-ITS2 area. Mycologia 1999, 91, 978–986. [Google Scholar] [CrossRef]
- Adamcik, S.; Ripkova, S. New collections of Flammulina rossica. Czech Mycol. 2008, 60, 113–121. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.; Blanchette, R.A.; Ander, P. Morphological aspects of wood degradation by fungi and bacteria. In Microbial and Ezymatic Degradation of Wood and Wood Components; Springer: Berlin/Heidelberg, Germany, 1990; pp. 1–87. [Google Scholar]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeong, Y.U.; Kong, W.S. Genome sequencing and carbohydrate-active enzyme (CAZyme) repertoire of the white rot fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef]
- Lee, C.S.; Kong, W.S.; Park, Y.J. Genome sequencing and genome-wide identification of carbohydrate-active enzymes (CAZymes) in the white rot fungus Flammulina fennae. Microbiol. Biotechnol. Lett. 2018, 46, 300–312. [Google Scholar] [CrossRef]
- Park, Y.J.; Kong, W.S. Genome-wide comparison of carbohydrate-active enzymes (CAZymes) repertoire of Flammulina ononidis. Mycobiology 2018, 46, 349–360. [Google Scholar] [CrossRef]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Sollewijn Gelpke, M.D.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.-J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 4 May 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for β short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef]
- Staats, M.; van Kan, J.A. Genome update of Botrytis cinerea strains B05. 10 and T4. Eukaryot. Cell 2012, 11, 1413–1414. [Google Scholar] [CrossRef]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagye, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C.K.; et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Aerts, A.; Ahrén, D.; Brun, A.; Danchin, E.G.J.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [PubMed]
- Janbon, G.; Ormerod, K.L.; Paulet, D.; Byrnes, E.J., III; Yadav, V.; Chatterjee, G.; Mullapudi, N.; Hon, C.-C.; Billmyre, R.B.; Brunel, F.; et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLOS Genet. 2014, 10, e1004261. [Google Scholar]
- Fisk, D.G.; Ball, C.A.; Dolinski, K.; Engel, S.R.; Hong, E.L.; Issel-Tarver, L.; Schwartz, K.; Sethuraman, A.; Botstein, D.; Cherry, J.M. Saccharomyces cerevisiae S288C genome annotation: A working hypothesis. Yeast 2006, 23, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Ohm, R.A.; De Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef]
- Li, W.C.; Huang, C.H.; Chen, C.L.; Chuang, Y.C.; Tung, S.Y.; Wang, T.F. Trichoderma reesei complete genome sequence, repeat-induced point mutation, and partitioning of CAZyme gene clusters. Biotechnol. Biofuels 2017, 10, 170. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.C.; Weinstein, J.; Ujita, E.L.; Riggs, K.J.; Lai, H. The membrane-binding domain of a rat liver Golgi sialyltransferase. Biochem. Soc. Trans. 1987, 15, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Wickner, W.T.; Lodish, H.F. Multiple mechanisms of protein insertion into and across membranes. Science 1985, 230, 400–407. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Biely, P. Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol. Adv. 2012, 30, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.M.; Nascimento, A.S.; Polikarpov, I. Structural diversity of carbohydrate esterases. Biotechnol. Res. Innov. 2017, 1, 35–51. [Google Scholar] [CrossRef]
- Adesioye, F.A.; Makhalanyane, T.P.; Biely, P.; Cowan, D.A. Phylogeny, classification and metagenomic bioprospecting of microbial acetyl xylan esterases. Enzyme. Microb. Technol. 2016, 93, 79–91. [Google Scholar] [CrossRef]
- Wei, Y.; Schottel, J.L.; Derewenda, U.; Swenson, L.; Patkar, S.; Derewenda, Z.S. A novel variant of the catalytic triad in the Streptomyces scabies esterase. Nat. Struct. Biol. 1995, 2, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.I.; Valinger, G.; Sölkner, B.; Stubenrauch, G.; Schwab, H. A novel esterase from Burkholderia gladioli shows high deacetylation activity on cephalosporins is related to β-lactamases and dd-peptidases. J. Biotechnol. 2001, 89, 11–25. [Google Scholar] [CrossRef]
- Christov, L.P.; Prior, B.A. Esterases of xylan-degrading microorganisms: Production, properties, and significance. Enzyme Microb. Tech. 1993, 15, 460–475. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Glycoside hydrolases across environmental microbial communities. PLoS Comput. Biol. 2016, 12, e1005300. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar] [CrossRef]
- Hahn, M.; Olsen, O.; Politz, O.; Borriss, R.; Heinemann, U. Crystal structure and site-directed mutagenesis of Bacillus macerans endo-1,3-1,4-β -glucanase. J. Biol. Chem. 1995, 270, 3081–3088. [Google Scholar] [CrossRef]
- Masuda, S.; Endo, K.; Koizumi, N.; Hayami, T.; Fukazawa, T.; Yatsunami, R.; Fukui, T.; Nakamura, S. Molecular identification of a novel β-1,3-glucanase from alkaliphilic Nocardiopsis sp. strain F96. Extremophiles 2006, 10, 251–255. [Google Scholar] [CrossRef]
- Kotake, T.; Hirata, N.; Degi, Y.; Ishiguro, M.; Kitazawa, K.; Takata, R.; Ichinose, H.; Kaneko, S.; Igarashi, K.; Samejima, M.; et al. Endo-β-1,3-galactanase from winter mushroom Flammulina velutipes. J. Biol. Chem. 2011, 286, 27848–27854. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Berlemont, R. Distribution and diversity of enzymes for polysaccharide degradation in fungi. Sci. Rep. 2017, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 2011, 14, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Berlemont, R.; Martiny, A.C. Phylogenetic distribution of potential cellulases in bacteria. Appl. Environ. Microbiol. 2013, 79, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Berlemont, R.; Martiny, A.C. Genomic potential for polysaccharides deconstruction in bacteria. Appl. Environ. Microbiol. 2015, 81, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Talamantes, D.; Biabini, N.; Dang, H.; Abdoun, K.; Berlemont, R. Natural diversity of cellulases, xylanases, and chitinases in bacteria. Biotechnol. Biofuels 2016, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Eichlerová, I.; Homolka, L.; Žifčáková, L.; Lisá, L.; Dobiášová, P.; Baldrian, P. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecol. 2015, 13, 10–22. [Google Scholar] [CrossRef]
- Treseder, K.K.; Lennon, J.T. Fungal traits that drive ecosystem dynamics on land. Microbiol. Mol. Biol. Rev. 2015, 79, 243–262. [Google Scholar] [CrossRef]
- Sutherland, I.W. Polysaccharide lyases. FEMS Microbiol. Rev. 1995, 16, 323–347. [Google Scholar] [CrossRef]
- Yip, V.L.; Withers, S.G. Breakdown of oligosaccharides by the process of elimination. Curr. Opin. Chem. Biol. 2006, 10, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction: Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Van den Brink, J.; de Vries, R.P. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477–1492. [Google Scholar] [CrossRef] [Green Version]
- Garron, M.L.; Cygler, M. Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 2010, 20, 1547–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhardt, R.J.; Galliher, P.M.; Cooney, C.L. Polysaccharide lyases. Appl. Biochem. Biotechnol. 1987, 12, 135–176. [Google Scholar] [CrossRef] [PubMed]
- Xavier-Santos, S.; Carvalho, C.C.; Bonfá, M.; Silva, R.; Capelari, M.; Gomes, E. Screening for pectinolytic activity of wood-rotting basidiomycetes and characterization of the enzymes. Folia Microbiol. (Praha) 2004, 49, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Reiss, R.; Ihssen, J.; Richter, M.; Eichhorn, E.; Schilling, B.; Thöny-Meyer, L. Laccase versus laccase-like multi-copper oxidase: A comparative study of similar enzymes with diverse substrate spectra. PLoS ONE 2013, 8, e65633. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.S.; Ruiz-Duenas, F.J.; Santillana, E.; Ferreira, P.; Martinez, M.J.; Martinez, A.T.; Romero, A. Novel structural features in the GMC family of oxidoreductases revealed by the crystal structure of fungal aryl-alcohol oxidase. Acta Crystallogr. 2009, D65, 1196–1205. [Google Scholar] [CrossRef]
- Wierenga, R.K.; Drenth, J.; Schulz, G.E. Comparison of the 3-dimensional protein and nucleotide structure of the FAD-binding domain of parahydroxybenzoate hydroxylase with the FAD-binding as well as NADPH-binding domains of glutathionereductase. J. Mol. Biol. 1983, 167, 725–739. [Google Scholar] [CrossRef]
- Varela, E.; Martinet, M.J.; Martinez, A.T. Arylalcohol oxidase protein sequence: A comparison with glucose oxidase and other FAD oxidoreductases. Biochem. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1481, 202–208. [Google Scholar] [CrossRef]
- Leonowicz, A.; Matuszewska, A.; Luterek, J.; Ziegenhagen, D.; Wojtas-Wasilewska, M.; Cho, N.S.; Hofrichter, M.; Rogalski, J. Biodegradation of lignin by white rot fungi. Fungal Genet. Biol. 1999, 27, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Guillén, F.; Martínez, M.J.; Gutiérrez, A.; Del Rio, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Shoseyov, O.; Shani, Z.; Levy, I. Carbohydrate binding modules: Biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 2006, 70, 283–295. [Google Scholar] [CrossRef] [PubMed]
- The CAZypedia Consortium. Ten years of CAZypedia: A living encyclopedia of carbohydrate-active enzymes. Glycobiology 2018, 28, 3–8. [Google Scholar]
- Várnai, A.; Mäkelä, M.R.; Djajadi, D.T.; Rahikainen, J.; Hatakka, A.; Viikari, L. Carbohydrate-binding modules of fungal cellulases: Occurrence in nature, function, and relevance in industrial biomass conversion. Adv. Appl. Microbiol. 2014, 88, 103–165. [Google Scholar] [PubMed]
- Mizuno, R.; Ichinose, H.; Honda, M.; Takabatake, K.; Sotome, I.; Takai, T.; Maehara, T.; Okadome, H.; Isobe, S.; Gau, M.; et al. Use of whole crop sorghums as a raw material in consolidated bioprocessing bioethanol production Using Flammulina velutipes. Biosci. Biotechnol. Biochem. 2009, 73, 1671–1673. [Google Scholar] [CrossRef]
- Mizuno, R.; Ichinose, H.; Maehara, T.; Takabatake, K.; Kaneko, S. Properties of ethanol fermentation by Flammulina velutipes. Biosci. Biotechnol. Biochem. 2009, 73, 2240–2245. [Google Scholar] [CrossRef]
- Lynd, L.R.; Van Zyl, W.H.; McBride, J.E.; Laser, M. Consolidated bioprocessing of cellulosic biomass: An update. Curr. Opin. Biotechnol. 2005, 16, 577–583. [Google Scholar] [CrossRef]
- Van Zyl, W.H.; Lynd, L.R.; Den Haan, R.; McBride, J.E. Consolidated bioprocessing for bioethanol production using Saccharomyces cerevisiae. Adv. Biochem. Eng. Biotechnol. 2007, 108, 205–235. [Google Scholar]
- Xu, Q.; Singh, A.; Himmel, M.E. Perspectives and new directions for the production of bioethanol using consolidated bioprocessing of lignocellulose. Curr. Opin. Biotechnol. 2009, 20, 364–371. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hiseq 2000 NGS Analysis | Total reads (100 bp) | 38,390,380 |
| Reads after trimming (%), >Q30 | 35,908,618 (93.53) | |
| Velvet De Novo Assembly | Optimized Velvet hash value (kmer) | 31 |
| Total number of contigs | 15,546 | |
| Number of contigs (>1 kb) | 1843 | |
| Contig N50 (bp) | 48,718 | |
| Length of longest contig (bp) | 418,162 | |
| Total bases in contigs (bp) | 35,645,506 | |
| Total bases in contigs (>1 kb) | 33,499,878 | |
| GC content (%) | 49.79 | |
| Gene Prediction | Predicted gene | 12,588 |
| Average gene length (bp) | 1911 | |
| Average protein length (aa) | 510.29 | |
| Average exon per gene | 6.52 | |
| Average exon size (bp) | 234.67 | |
| Average intron size (bp) | 68.03 |
| Fungal Species | Flammulina rossica | F. ononidis | F. fennae | F. elastica | F. velutipes | Laccaria bicolar | Criptocuccus cinerea | Phanerochaete chrysosporium | Schizophyllum commune |
| Strain | ASI4194 | KACC46186 | KACC46185 | KACC46182 | KACC42780 | S238N-H82 | Okayama7#130 | RP78 | H4-8 |
| Genome (Mb) | 35.6 | 34.5 | 32.4 | 35 | 35.6 | 64.9 | 37.5 | 35.1 | 38.5 |
| Genes | 12,588 | 12,269 | 11,591 | 12,536 | 12,218 | 20,614 | 13,544 | 10,048 | 13,181 |
| GC (%) | 49.79 | 49.76 | 39 | 49.7 | 48.99 | 46.6 | 51.6 | 53.2 | 56.6 |
| Average Gene Length (bp) | 1911 | 2009 | 1980 | 1973 | 2294 | 1533.0 | 1679.0 | 1667.0 | 1794.9 |
| Average Exon Size (bp) | 234.67 | 234.09 | 230.53 | 233.91 | 231.4 | 210.1 | 251.0 | 232.0 | 249.3 |
| Average Intron Size (bp) | 68.03 | 68.14 | 68.93 | 69.29 | 190.3 | 92.7 | 75.0 | 117.0 | 79.0 |
| Fungal Species | Genes | Genes in Orthogroups (%) | Unassigned Genes (%) | Orthogroups Containing Species (%) | Number of Species-Specific Orthogroups | Genes in Species-Specific Orthogroups |
|---|---|---|---|---|---|---|
| Flammulina rossica ASI4194 * | 12,588 | 12,024 (95.5) | 564 (4.5) | 7485 (58.7) | 5 | 12 |
| Flammulina fennae KACC46185 * | 11,591 | 11,235 (96.9) | 356 (3.1) | 7307 (57.3) | 2 | 4 |
| Flammulina ononidis KACC46186 * | 12,269 | 11,881 (96.8) | 388 (3.2) | 7572 (59.4) | 1 | 2 |
| Flammulina elastica KACC46182 * | 12,536 | 11,987 (95.6) | 549 (4.4) | 7592 (59.5) | 1 | 2 |
| Flammulina velutipes KACC42780 * | 12,218 | 10,774 (88.2) | 1444 (11.8) | 6679 (52.4) | 4 | 10 |
| Agaricus bisporus var. bisporus H97 * | 10,448 | 9102 (87.1) | 1346 (12.9) | 5712 (44.8) | 29 | 265 |
| Coprinopsis cinerea okayama 7#130 * | 13,356 | 10,837 (81.1) | 2519 (18.9) | 6393 (50.1) | 59 | 329 |
| Cryptococcus neoformans var. grubii H99 * | 7826 | 6477 (82.8) | 1349 (17.2) | 4719 (37.0) | 39 | 129 |
| Laccaria bicolor S238N-H82 * | 18,215 | 12,662 (69.5) | 5553 (30.5) | 6217 (48.7) | 73 | 524 |
| Lentinula edodes * | 14,079 | 11,678 (82.9) | 2401 (17.1) | 6674 (52.3) | 61 | 340 |
| Phanerochaete chrysosporium RP78 * | 13,602 | 10,443 (76.8) | 3159 (23.2) | 6223 (48.8) | 36 | 265 |
| Schizophyllum commune H4-8 * | 13,194 | 10,976 (83.2) | 2218 (16.8) | 6214 (48.7) | 59 | 437 |
| Aspergillus nidulans FGSC-A4 ** | 9561 | 8433 (88.2) | 1128 (11.8) | 5709 (44.8) | 4 | 24 |
| Botrytis cinerea B05.10 ** | 16,389 | 9336 (57.0) | 7053 (43.0) | 6263 (49.1) | 17 | 64 |
| Cordyceps militaris CM01 ** | 9651 | 8369 (86.7) | 1282 (13.3) | 6131 (48.1) | 5 | 13 |
| Neurospora crassa OR74A ** | 10,812 | 8659 (80.1) | 2153 (19.9) | 6217 (48.7) | 10 | 26 |
| Saccharomyces cerevisiae S288C ** | 6002 | 4624 (77.0) | 1378 (23.0) | 3485 (27.3) | 12 | 33 |
| Trichoderma reesei QM6a ** | 9115 | 8332 (91.4) | 783 (8.6) | 6283 (49.3) | 4 | 9 |
| Fungal Species | CAZymes | No. of CAZyme (Annotation DB) | Total | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| AA 1 | GH 2 | GT 3 | CE 4 | CBM 5 | PL 6 | ||||
| Flammulina rossica * | 93 | 182 | 64 | 56 | 7 | 17 | 419 (Hmmer dbCAN) | 528 | This study |
| 61 | 145 | 53 | 21 | 5 | 15 | 300 (Hotpep) | |||
| 39 | 155 | 72 | 11 | 11 | 6 | 294 (CAZy database) | |||
| Flammulina fennae * | 86 | 220 | 85 | 57 | 45 | 20 | - | 513 | [10] |
| Flammulina ononidis * | 87 | 228 | 87 | 61 | 40 | 21 | - | 524 | [11] |
| Flammulina elastica * | 82 | 218 | 89 | 59 | 42 | 18 | - | 508 | [9] |
| Flammulina velutipes * | 85 | 239 | 84 | 63 | 44 | 25 | - | 540 | [9] |
| Agaricus bisporus * | 81 | 174 | 54 | 33 | 44 | 9 | - | 395 | JGI database |
| Cryptococcus cinerea * | 111 | 195 | 83 | 60 | 105 | 16 | - | 570 | [9] |
| Laccaria bicolor * | 55 | 170 | 96 | 18 | 31 | 7 | - | 377 | JGI database |
| Lentinula edodes * | 82 | 254 | 85 | 44 | 61 | 11 | - | 537 | [9] |
| Phanerochaete chrysosporium * | 85 | 175 | 65 | 16 | 62 | 4 | - | 407 | JGI database |
| Schizophyllum commune * | 78 | 241 | 85 | 57 | 37 | 18 | - | 516 | [9] |
| Cryptococcus neoformans * | 14 | 97 | 70 | 5 | 12 | 4 | - | 202 | CAZy database |
| Cordyceps militaris ** | 54 | 165 | 91 | 34 | 39 | 5 | - | 388 | [9] |
| Trichoderma reesei ** | 59 | 210 | 90 | 32 | 44 | 5 | - | 440 | [9] |
| Saccharomyces cerevisiae ** | 5 | 57 | 68 | 2 | 12 | 0 | - | 144 | CAZy database |
| Aspergillus nidulans ** | 33 | 267 | 91 | 30 | 46 | 23 | - | 490 | CAZy database |
| Neurospora crassa ** | 35 | 177 | 80 | 21 | 42 | 4 | - | 359 | CAZy database |
| Botrytis cinerea ** | 77 | 287 | 119 | 37 | 89 | 10 | - | 619 | CAZy database |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.-J.; Lee, C.-S.; Kong, W.-S. Genomic Insights into the Fungal Lignocellulolytic Machinery of Flammulina rossica. Microorganisms 2019, 7, 421. https://doi.org/10.3390/microorganisms7100421
Park Y-J, Lee C-S, Kong W-S. Genomic Insights into the Fungal Lignocellulolytic Machinery of Flammulina rossica. Microorganisms. 2019; 7(10):421. https://doi.org/10.3390/microorganisms7100421
Chicago/Turabian StylePark, Young-Jin, Chang-Soo Lee, and Won-Sik Kong. 2019. "Genomic Insights into the Fungal Lignocellulolytic Machinery of Flammulina rossica" Microorganisms 7, no. 10: 421. https://doi.org/10.3390/microorganisms7100421