Reduced Virulence of an Introduced Forest Pathogen over 50 Years

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. New Zealand Dothistroma septosporum Isolates, Growth, and Dothistromin Assays
2.2. DNA Sequencing and Read Mapping
2.3. Single Nucleotide Polymorphism Analysis
2.4. Mutation Rate Estimation
2.5. Pathogenicity Assays
2.6. Spore Germination, Growth, and Copper Tolerance Assays
2.7. Statistical Analysis
3. Results
3.1. Genome Sequencing and Single Nucleotide Polymorphisms of D. septosporum Isolates
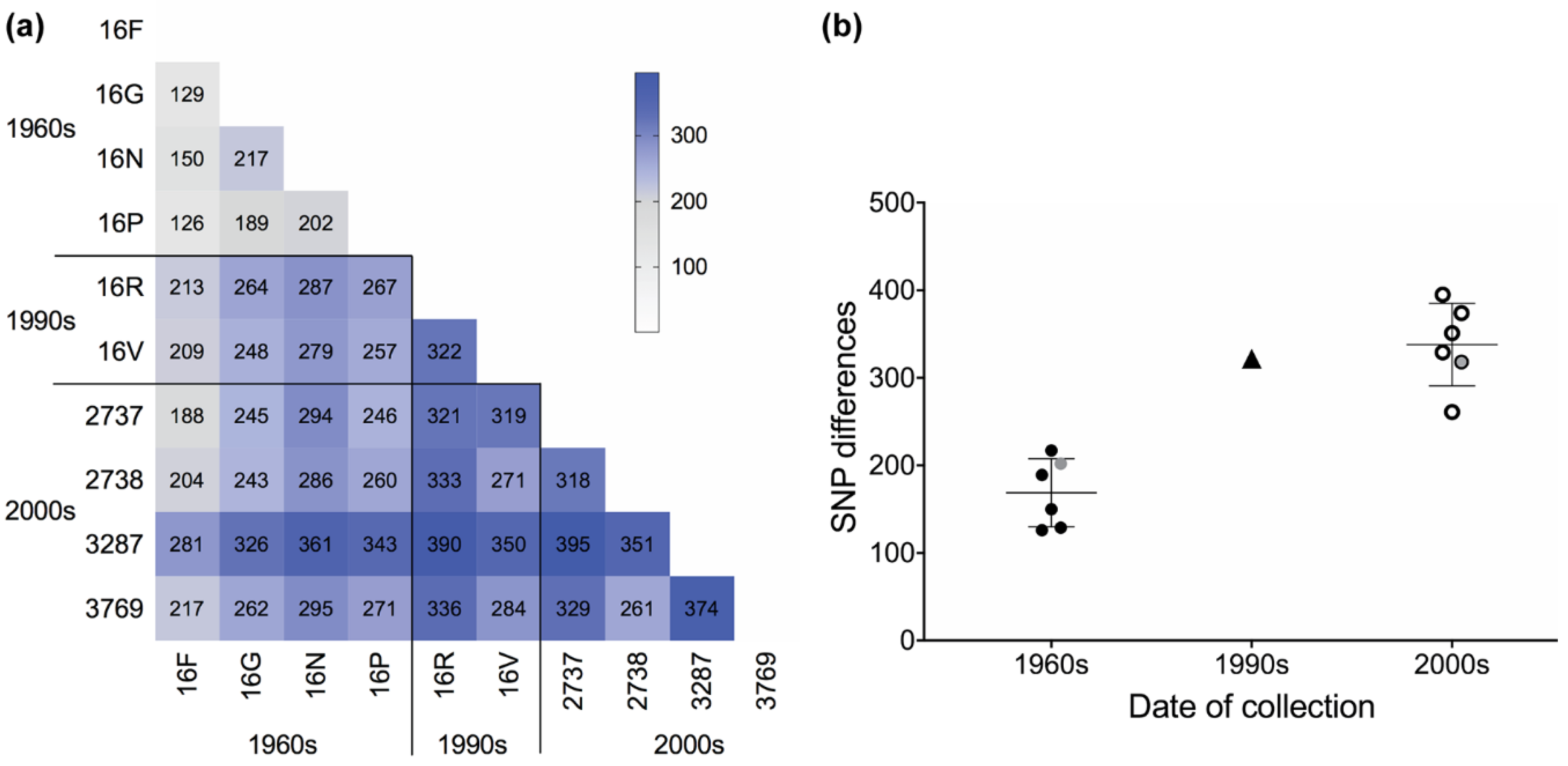
3.2. SNP Differences Reveal Increased Diversity between Isolates over Time
3.3. Estimating Mutation Rates
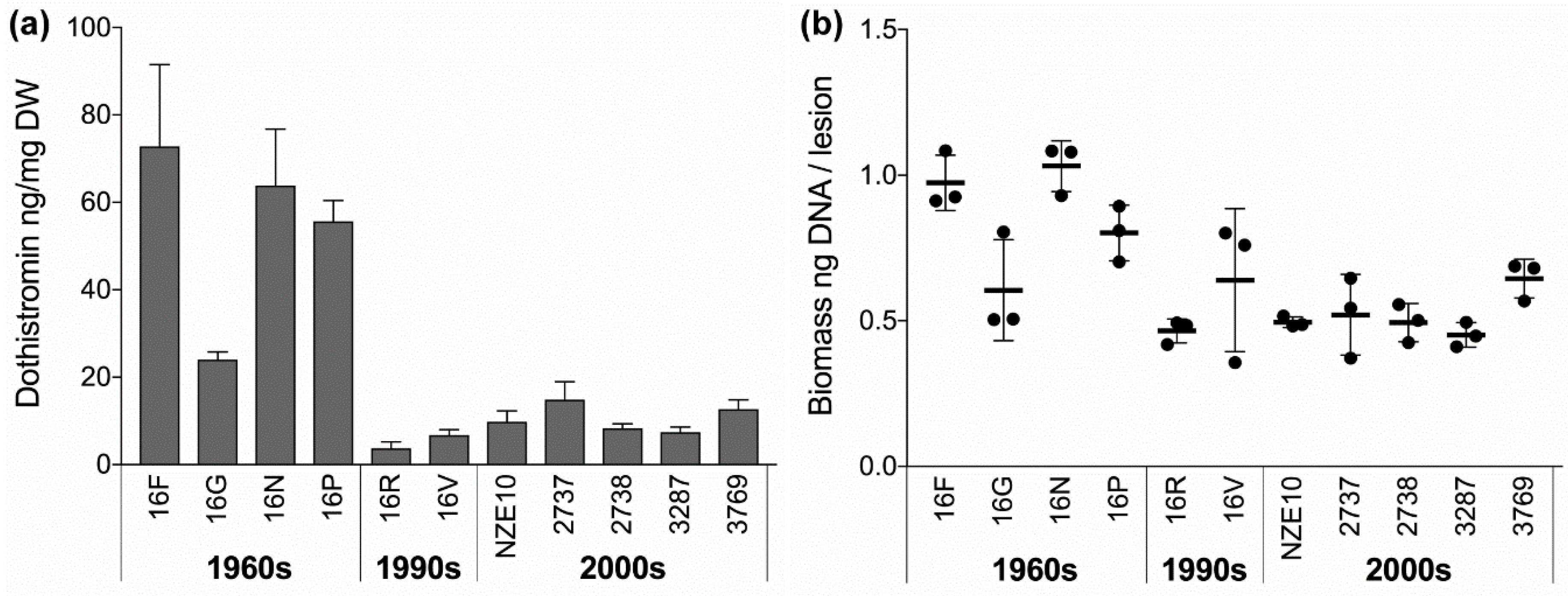
3.4. Dothistromin Production and Virulence Declined Significantly over Time
3.5. Sporulation, Growth, and the Effects of Copper
3.6. Are Specific Genetic Changes Associated with Differences in Phenotypes?
4. Discussion
4.1. Genome Diversity Suggests the Introduction of Several Closely-Related Isolates of D. septosporum
4.2. Estimation of a Mutation Rate
4.3. Spore Germination, Growth Rates, and Copper Effects
4.4. Virulence and Dothistromin Production Decreased Over Time
4.5. A Genetic Basis for Changes in Dothistromin Production and Virulence Could Not Be Determined
4.6. Was There a Trade-Off between Virulence and Pathogen Transmission?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ussiri, D.A.; Lal, R. Global forests management for climate change mitigation. In Carbon Sequestration for Climate Change Mitigation and Adaptation; Springer: Berlin/Heidelberg, Germany, 2017; pp. 395–432. [Google Scholar]
- Law, B.E.; Hudiburg, T.W.; Berner, L.T.; Kent, J.J.; Buotte, P.C.; Harmon, M.E. Land use strategies to mitigate climate change in carbon dense temperate forests. Proc. Natl. Acad. Sci. USA 2018, 115, 3663. [Google Scholar] [CrossRef]
- Woods, A. Is the health of British Columbia’s forests being influenced by climate change? If so, was this predictable? Can. J. Plant Pathol. 2011, 33, 117–126. [Google Scholar] [CrossRef]
- Jung, T.; Orlikowski, L.; Henricot, B.; Abad-Campos, P.; Aday, A.; Aguín Casal, O.; Bakonyi, J.; Cacciola, S.; Cech, T.; Chavarriaga, D. Widespread Phytophthora infestations in European nurseries put forest, semi-natural and horticultural ecosystems at high risk of Phytophthora diseases. For. Pathol. 2016, 46, 134–163. [Google Scholar] [CrossRef]
- Brasier, C.M. The biosecurity threat to the UK and global environment from international trade in plants. Plant Pathol. 2008, 57, 792–808. [Google Scholar] [CrossRef]
- Ghelardini, L.; Pepori, A.L.; Luchi, N.; Capretti, P.; Santini, A. Drivers of emerging fungal diseases of forest trees. For. Ecol. Manag. 2016, 381, 235–246. [Google Scholar] [CrossRef]
- Gonthier, P.; Giordano, L.; Nicolotti, G. Further observations on sudden diebacks of Scots pine in the European Alps. Forest. Chron. 2010, 86, 110–117. [Google Scholar] [CrossRef]
- Desprez-Loustau, M.-L.; Aguayo, J.; Dutech, C.; Hayden, K.J.; Husson, C.; Jakushkin, B.; Marcais, B.; Piou, D.; Robin, C.; Vacher, C. An evolutionary ecology perspective to address forest pathology challenges of today and tomorrow. Ann. Forest Sci. 2016, 73, 45–67. [Google Scholar] [CrossRef] [Green Version]
- Wingfield, M.J.; Brockerhoff, E.G.; Wingfield, B.D.; Slippers, B. Planted forest health: The need for a global strategy. Science 2015, 349, 832–836. [Google Scholar] [CrossRef]
- Bulman, L.S.; Bradshaw, R.E.; Fraser, S.; Martín-García, J.; Barnes, I.; Musolin, D.L.; La Porta, N.; Woods, A.J.; Diez, J.J.; Koltay, A.; et al. A worldwide perspective on the management and control of Dothistroma needle blight. For. Pathol. 2016, 46, 472–488. [Google Scholar] [CrossRef] [Green Version]
- Plissonneau, C.; Benevenuto, J.; Mohd-Assaad, N.; Fouche, S.; Hartmann, F.E.; Croll, D. Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front. Plant Sci. 2017, 8, 119. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; McDonald, B.A. The origins of plant pathogens in agro-ecosystems. Annu. Rev. Phytopathol. 2008, 46, 75–100. [Google Scholar] [CrossRef]
- Jeger, M.J.; Pautasso, M. Plant disease and global change–the importance of long-term data sets. New Phytol. 2008, 177, 8–11. [Google Scholar] [CrossRef]
- Bearchell, S.J.; Fraaije, B.A.; Shaw, M.W.; Fitt, B.D.L. Wheat archive links long-term fungal pathogen population dynamics to air pollution. Proc. Natl. Acad. Sci. USA 2005, 102, 5438–5442. [Google Scholar] [CrossRef] [Green Version]
- Kinloch, B.B.; Davis, D.A.; Burton, D. Resistance and virulence interactions between two white pine species and blister rust in a 30-year field trial. Tree Genet. Genom. 2008, 4, 65–74. [Google Scholar] [CrossRef]
- Woods, A.J.; Martín-García, J.; Bulman, L.; Vasconcelos, M.W.; Boberg, J.; La Porta, N.; Peredo, H.; Vergara, G.; Ahumada, R.; Brown, A.; et al. Dothistroma needle blight, weather and possible climatic triggers for the disease’s recent emergence. For. Pathol. 2016, 46, 443–452. [Google Scholar] [CrossRef]
- Gonthier, P.; Nicolotti, G.; Linzer, R.; Guglielmo, F.; Garbelotto, M. Invasion of European pine stands by a North American forest pathogen and its hybridization with a native interfertile taxon. Mol. Ecol. 2007, 16, 1389–1400. [Google Scholar] [CrossRef]
- Gonthier, P.; Garbelotto, M. Amplified fragment length polymorphism and sequence analyses reveal massive gene introgression from the European fungal pathogen Heterobasidion annosum into its introduced congener H. irregulare. Mol. Ecol. 2011, 20, 2756–2770. [Google Scholar] [CrossRef]
- Garbelotto, M.; Guglielmo, F.; Mascheretti, S.; Croucher, P.; Gonthier, P. Population genetic analyses provide insights on the introduction pathway and spread patterns of the North American forest pathogen Heterobasidion irregulare in Italy. Mol. Ecol. 2013, 22, 4855–4869. [Google Scholar] [CrossRef]
- Drenkhan, R.; Tomešová-Haataja, V.; Fraser, S.; Bradshaw, R.E.; Vahalík, P.; Mullett, M.S.; Martín-García, J.; Bulman, L.S.; Wingfield, M.J.; Kirisits, T.; et al. Global geographic distribution and host range of Dothistroma species: A comprehensive review. For. Pathol. 2016, 46, 408–442. [Google Scholar] [CrossRef]
- Groenewald, M.; Barnes, I.; Bradshaw, R.E.; Brown, A.V.; Dale, A.; Groenewald, J.Z.; Lewis, K.J.; Wingfield, B.D.; Wingfield, M.J.; Crous, P.W. Characterization and distribution of mating type genes in the Dothistroma needle blight pathogens. Phytopathology 2007, 97, 825–834. [Google Scholar] [CrossRef]
- Barnes, I.; Wingfield, M.J.; Carbone, I.; Kirisits, T.; Wingfield, B.D. Population structure and diversity of an invasive pine needle pathogen reflects anthropogenic activity. Ecol. Evol. 2014, 4, 3642–3661. [Google Scholar] [CrossRef] [PubMed]
- Hirst, P.; Richardson, T.E.; Carson, S.D.; Bradshaw, R.E. Dothistroma pini genetic diversity is low in New Zealand. N. Z. J. For. Sci. 1999, 29, 459–472. [Google Scholar]
- Bradshaw, R.E.; Sim, A.D.; Chettri, P.; Dupont, P.-Y.; Guo, Y.; Hunziker, L.; McDougal, R.L.; Van der Nest, A.; Fourie, A.; Wheeler, D.; et al. Global population genomics of the forest pathogen Dothistroma septosporum reveal chromosome duplications in high dothistromin-producing strains. Mol. Plant Pathol. 2019, 20, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Barnes, I.; Crous, P.W.; Wingfield, B.D.; Wingfield, M.J. Multigene phylogenies reveal that red band needle bight of Pinus is caused by two distinct species of Dothistroma, D. septosporum and D. pini. Stud. Mycol. 2004, 50, 551–565. [Google Scholar]
- de Wit, P.J.G.M.; van der Burgt, A.; Okmen, B.; Stergiopoulos, I.; Abd-Elsalam, K.A.; Aerts, A.L.; Bahkali, A.H.; Beenen, H.G.; Chettri, P.; Cox, M.P.; et al. The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012, 8, e1003088. [Google Scholar] [CrossRef]
- Chettri, P.; Calvo, A.M.; Cary, J.W.; Dhingra, S.; Guo, Y.A.; McDougal, R.L.; Bradshaw, R.E. The veA gene of the pine needle pathogen Dothistroma septosporum regulates sporulation and secondary metabolism. Fungal Genet. Biol. 2012, 49, 141–151. [Google Scholar] [CrossRef]
- Aronesty, E. Ea-Utils: Command-Line Tools for Processing Biological Sequencing Data. Available online: https://expressionanalysis.github.io/ea-utils/ (accessed on 3 May 2018).
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Bioinformatics, B. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2011; Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 May 2018).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army tool for genome feature analysis. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; pp. 11–12. [Google Scholar]
- Bradshaw, R.E.; Guo, Y.; Sim, A.D.; Kabir, M.S.; Chettri, P.; Ozturk, I.K.; Hunziker, L.; Ganley, R.J.; Cox, M.P. Genome-wide gene expression dynamics of the fungal pathogen Dothistroma septosporum throughout its infection cycle of the gymnosperm host Pinus radiata. Mol. Plant Pathol. 2016, 17, 210–224. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McDougal, R.L.; Yang, S.; Schwelm, A.; Stewart, A.; Bradshaw, R.E. A novel GFP-based approach for screening biocontrol microorganisms in vitro against Dothistroma septosporum. J. Microbiol. Methods 2011, 87, 32–37. [Google Scholar] [CrossRef]
- Kabir, M.S.; Ganley, R.J.; Bradshaw, R.E. An improved artificial pathogenicity assay for Dothistroma needle blight on Pinus radiata. Aust. Plant Pathol. 2013, 42, 503–510. [Google Scholar] [CrossRef]
- Olsen, P.F. Dothistroma spraying experience-1966 to 1970. N. Z. J. For. 1971, 16, 101–104. [Google Scholar]
- Bulman, L.; Gadgil, P.; Kershaw, D.; Ray, J. Assessment and control of Dothistroma needle blight. For. R. N. Z. For. Res. Bull. No. 229 2004. [Google Scholar]
- Bradshaw, R.E.; Ganley, R.J.; Jones, W.T.; Dyer, P.S. High levels of dothistromin toxin produced by the forest pathogen Dothistroma pini. Mycol. Res. 2000, 104, 325–332. [Google Scholar] [CrossRef]
- R Development Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.r-project.org (accessed on 20 April 2018).
- Hayes, W.L. Statistics for Psychologists; Holt, Rinehart and Winston Inc.: New York, NY, USA, 1963; p. 325. [Google Scholar]
- Lüdecke, D. Sjstats: Statistical Functions for Regression Models (Version 0.17.2). 2019. Available online: https://CRAN.R-project.org/package=sjstats (accessed on 20 April 2018).
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; Team, R.C. Nlme: Linear and Nonlinear Mixed Effects Models (Version 3.1-137). 2019. Available online: https://CRAN.R-project.org/package=nlme (accessed on 20 April 2018).
- Kabir, M.S.; Ganley, R.J.; Bradshaw, R.E. Dothistromin toxin is a virulence factor in Dothistroma needle blight of pines. Plant Pathol. 2015, 64, 225–234. [Google Scholar] [CrossRef]
- Chettri, P.; Ehrlich, K.C.; Cary, J.W.; Collemare, J.; Cox, M.P.; Griffiths, S.A.; Olson, M.A.; de Wit, P.J.G.M.; Bradshaw, R.E. Dothistromin genes at multiple separate loci are regulated by AflR. Fungal Genet. Biol. 2013, 51, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, R.E.; Feng, Z.; Schwelm, A.; Yang, Y.; Zhang, S. Functional analysis of a putative dothistromin toxin MFS transporter gene. Toxins 2009, 1, 173–187. [Google Scholar] [CrossRef]
- Chettri, P.; Bradshaw, R.E. LaeA negatively regulates dothistromin production in the pine needle pathogen Dothistroma septosporum. Fungal Genet. Biol. 2016, 97, 24–32. [Google Scholar] [CrossRef]
- Chettri, P.; Dupont, P.-Y.; Bradshaw, R.E. Chromatin-level regulation of the fragmented dothistromin gene cluster in the forest pathogen Dothistroma septosporum. Mol. Microbiol. 2018, 107, 508–522. [Google Scholar] [CrossRef]
- Gilmour, J.W. Distribution and significance of the needle blight of pines caused by Dothistroma pini in New Zealand. Plant Dis. Rep. 1967, 51, 727–730. [Google Scholar]
- Kasuga, T.; White, T.J.; Taylor, J.W. Estimation of nucleotide substitution rates in Eurotiomycete fungi. Mol. Biol. Evol. 2002, 19, 2318–2324. [Google Scholar] [CrossRef]
- Zhu, Y.O.; Siegal, M.L.; Hall, D.W.; Petrov, D.A. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. USA 2014, 111, E2310–E2318. [Google Scholar] [CrossRef] [Green Version]
- Telfer, E.; Graham, N.; Macdonald, L.; Sturrock, S.; Wilcox, P.; Stanbra, L. Approaches to variant discovery for conifer transcriptome sequencing. PLoS ONE 2018, 13, e0205835. [Google Scholar] [CrossRef] [PubMed]
- Stoessl, A.; Abramowski, Z.; Lester, H.H.; Rock, G.L.; Towers, G.H.N. Further toxic properties of the fungal metabolite dothistromin. Mycopathologia 1990, 112, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Youngman, R.J.; Elstner, E.F. Photodynamic and reductive mechanisms of oxygen activation by the fungal phytotoxins, cercosporin and dothistromin. In Oxygen Radicals in Chemistry and Biology; Bors, W., Saran, M., Eds.; Walter de Gruyter and Co.: Berlin, NY, USA, 1984; pp. 501–508. [Google Scholar]
- Strobel, G.A. The emergence of endophytic microbes and their biological promise. J. Fungi 2018, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Pais, M.; Yoshida, K.; Giannakopoulou, A.; Pel, M.A.; Cano, L.M.; Oliva, R.F.; Witek, K.; Lindqvist-Kreuze, H.; Vleeshouwers, V.; Kamoun, S. Gene expression polymorphism underpins evasion of host immunity in an asexual lineage of the Irish potato famine pathogen. BMC Evol. Biol. 2018, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Gladieux, P.; Feurtey, A.; Hood, M.E.; Snirc, A.; Clavel, J.; Dutech, C.; Roy, M.; Giraud, T. The population biology of fungal invasions. Mol. Ecol. 2015, 24, 1969–1986. [Google Scholar] [CrossRef] [PubMed]
- Ganley, R.J.; Newcombe, G. Fungal endophytes in seeds and needles of Pinus monticola. Mycol. Res. 2006, 110, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Glen, M.; McDougal, R. Molecular tools for differentiating Cyclaneusma minus morphotypes and assessing their distribution in Pinus radiata forests in New Zealand. N. Z. J. For. Sci. 2016, 46, 21. [Google Scholar] [CrossRef]
- Nelson, P.G.; May, G. Coevolution between mutualists and parasites in symbiotic communities may lead to the evolution of lower virulence. Am. Nat. 2017, 190, 803–817. [Google Scholar] [CrossRef]
- Baalen, M.v.; Sabelis, M.W. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 1995, 146, 881–910. [Google Scholar] [CrossRef]
- Rigling, D.; Prospero, S. Cryphonectria parasitica, the causal agent of chestnut blight: Invasion history, population biology and disease control. Mol. Plant Pathol. 2018, 19, 7–20. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Yang, N.; Wen, Z.; Sun, X.; Chai, Y.; Ma, Z. Wheat microbiome bacteria can reduce virulence of a plant pathogenic fungus by altering histone acetylation. Nat. Commun. 2018, 9, 3429. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Isolate a | Year of Collection | Pinus Host Species | Region b | Mapped Reads c | % Mapped Reads c | SRA Accession Number d | Number of SNPs e |
|---|---|---|---|---|---|---|---|
| NZE10 | 2005 | P. radiata | Westland SI | n/a | n/a | PRJNA74753 | n/a |
| 16F | 1965 | P. contorta | Taupo | 48490708 | 94.2 | SRX3493225 | 227 |
| 16G | 1967 | P. menziensii | Taupo | 43651635 | 93.1 | SRX3493226 | 274 |
| 16N * | 1969 | P. radiata | Bay of Plenty | 46614557 | 94.5 | SRX3493227 | 311 |
| 16P * | 1969 | P. radiata | Bay of Plenty | 46230530 | 95.1 | SRX3493228 | 279 |
| 16R | 1991 | P. radiata | Waikato | 51420587 | 95.3 | SRX3493230 | 356 |
| 16V | 1994 | P. radiata | Bay of Plenty | 49590713 | 94.9 | SRX3493229 | 350 |
| 2737 * | 2006 | P. radiata | Taupo | 46549085 | 93.8 | SRX3493231 | 335 |
| 2738 * | 2006 | P. radiata | Taupo | 45942540 | 94.4 | SRX3493232 | 343 |
| 3287 | 2007 | P. radiata | Taupo | 47062342 | 93.9 | SRX3493234 | 424 |
| 3769 | 2013 | P. radiata | Gisborne | 46616890 | 93.9 | SRX3493233 | 362 |
| 4554 f | 2017 | P. yunnanensis | Bay of Plenty | n/a | n/a | n/a | n/a |
| Phenotype a | Decade Group (Mean ± SD) b | All Groups Comparison c | 1960s vs. 2000s Comparison c | ||||
|---|---|---|---|---|---|---|---|
| 1960s | 1990s | 2000s | ω2 | p Value | ω2 | p Value | |
| Dothistromin ng/mg DW | 54.10 ± 21.65 | 5.22 ± 2.07 | 10.62 ± 3.54 | 0.74 | 0.002 | 0.70 | 0.003 |
| Growth 1 (mm/day) 0 Cu | 0.61 ± 0.08 | 0.70 ± 0.17 | 0.69 ± 0.05 | 0.18 | 0.393 | 0.31 | 0.090 |
| Growth 1 (mm/day) 50 Cu | 0.56 ± 0.07 | 0.54 ± 0.10 | 0.57 ± 0.04 | 0.01 | 0.858 | 0.01 | 0.698 |
| Growth 1 ratio 50/0 Cu | 0.93 ± 0.06 | 0.79 ± 0.16 | 0.80 ± 0.07 | 0.33 | 0.018 | 0.49 | 0.003 |
| Growth 2 (mm/day) 0 Cu | 0.67 ± 0.09 | 0.77 ± 0.06 | 0.74 ± 0.12 | 0.14 | 0.434 | 0.11 | 0.322 |
| Growth 2 (mm/day) 100 Cu | 0.53 ± 0.04 | 0.60 ± 0.06 | 0.62 ± 0.05 | 0.41 | 0.033 | 0.47 | 0.006 |
| Growth 2 ratio 100/0 Cu | 0.81 ± 0.10 | 0.80 ± 0.13 | 0.84 ± 0.14 | 0.04 | 0.747 | 0.03 | 0.535 |
| Sporulation × 106/mL | 5.25 ± 1.91 | 2.70 ± 1.56 | 5.31 ± 4.91 | 0.05 | 0.646 | −0.01 | 0.979 |
| % spore germination 0 Cu | 35.79 ± 5.40 | 42.00 ± 11.15 | 44.52 ± 8.85 | 0.14 | 0.014 | 0.22 | 0.008 |
| % spore germination 50 Cu | 18.17 ± 5.66 | 13.33 ± 4.09 | 18.11 ± 5.77 | 0.05 | 0.187 | −0.03 | 0.704 |
| Spore germination ratio 50/0 Cu | 0.49 ± 0.17 | 0.32 ± 0.11 | 0.43 ± 0.17 | 0.07 | 0.140 | 0.00 | 0.349 |
| Disease lesions/100 needles | 16.42 ± 10.39 | 21.33 ± 25.29 | 18.92 ± 14.52 | −0.03 | 0.754 | −0.01 | 0.558 |
| Biomass in planta ng DNA/lesion | 0.85 ± 0.20 | 0.55 ± 0.18 | 0.52 ± 0.09 | 0.50 | 0.016 | 0.55 | 0.009 |
| Biomass in planta ng DNA/mg DW | 1.51 ± 0.54 | 0.84 ± 0.14 | 0.82 ± 0.30 | 0.43 | 0.061 | 0.41 | 0.040 |
| Gene | 16F | 16G | 16N | 16P | 16R | 16V | 2737 | 2738 | 3287 | 3769 | NZE10 | Predicted Function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 29186 | SN | SN | SN | SN | Glycoside hydrolase 72 | |||||||
| 33501 | SN | SN | ATP binding | |||||||||
| 34615 | SN | SN | SN | Not known | ||||||||
| 38669 | R113Q | R113Q | R113Q | R113Q | Not known | |||||||
| 54626 | G579V | G579V | transporter | |||||||||
| 71301 | SN | SN | SN | SN | ER protein | |||||||
| 71790 | G237D | G237D | Chromatin remodelling | |||||||||
| 73448 | I658V | I658V | I658V | I658V | Ubiquitin ligase | |||||||
| 74128 | A17G | A17G | Nucleoside metabolism | |||||||||
| 74795 | T482I | T482I | T482I | Vesicular transport | ||||||||
| 79913 | Q212P | Q212P | Q212P | Q212P | Lipid metabolism | |||||||
| 71010 | I493F | R665W | FS 329 | C570Y | Transcription regulation | |||||||
| 72963 | P456L | V728M | Chromatin remodelling | |||||||||
| 74323 | C319* | E643G | FS144 | S636* | FS 48 | FS 547 | RNA splicing | |||||
| 137952 | C92R | I222M, N226H | L327R, S328P | RNA processing |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradshaw, R.E.; Ormond, S.; Dupont, P.-Y.; Chettri, P.; Ozturk, I.K.; McDougal, R.L.; Bulman, L.S.; Cox, M.P. Reduced Virulence of an Introduced Forest Pathogen over 50 Years. Microorganisms 2019, 7, 420. https://doi.org/10.3390/microorganisms7100420
Bradshaw RE, Ormond S, Dupont P-Y, Chettri P, Ozturk IK, McDougal RL, Bulman LS, Cox MP. Reduced Virulence of an Introduced Forest Pathogen over 50 Years. Microorganisms. 2019; 7(10):420. https://doi.org/10.3390/microorganisms7100420
Chicago/Turabian StyleBradshaw, Rosie E., Shannon Ormond, Pierre-Yves Dupont, Pranav Chettri, I. Kutay Ozturk, Rebecca L. McDougal, Lindsay S. Bulman, and Murray P. Cox. 2019. "Reduced Virulence of an Introduced Forest Pathogen over 50 Years" Microorganisms 7, no. 10: 420. https://doi.org/10.3390/microorganisms7100420