
From Investigating a Case of Cellulitis to Exploring Nosocomial Infection Control of ST1 Legionella pneumophila Using Genomic Approaches
 , , , , , and
, , , , , and
Abstract
:1. Case Reports/Background
1.1. Case 1
1.2. Case 2
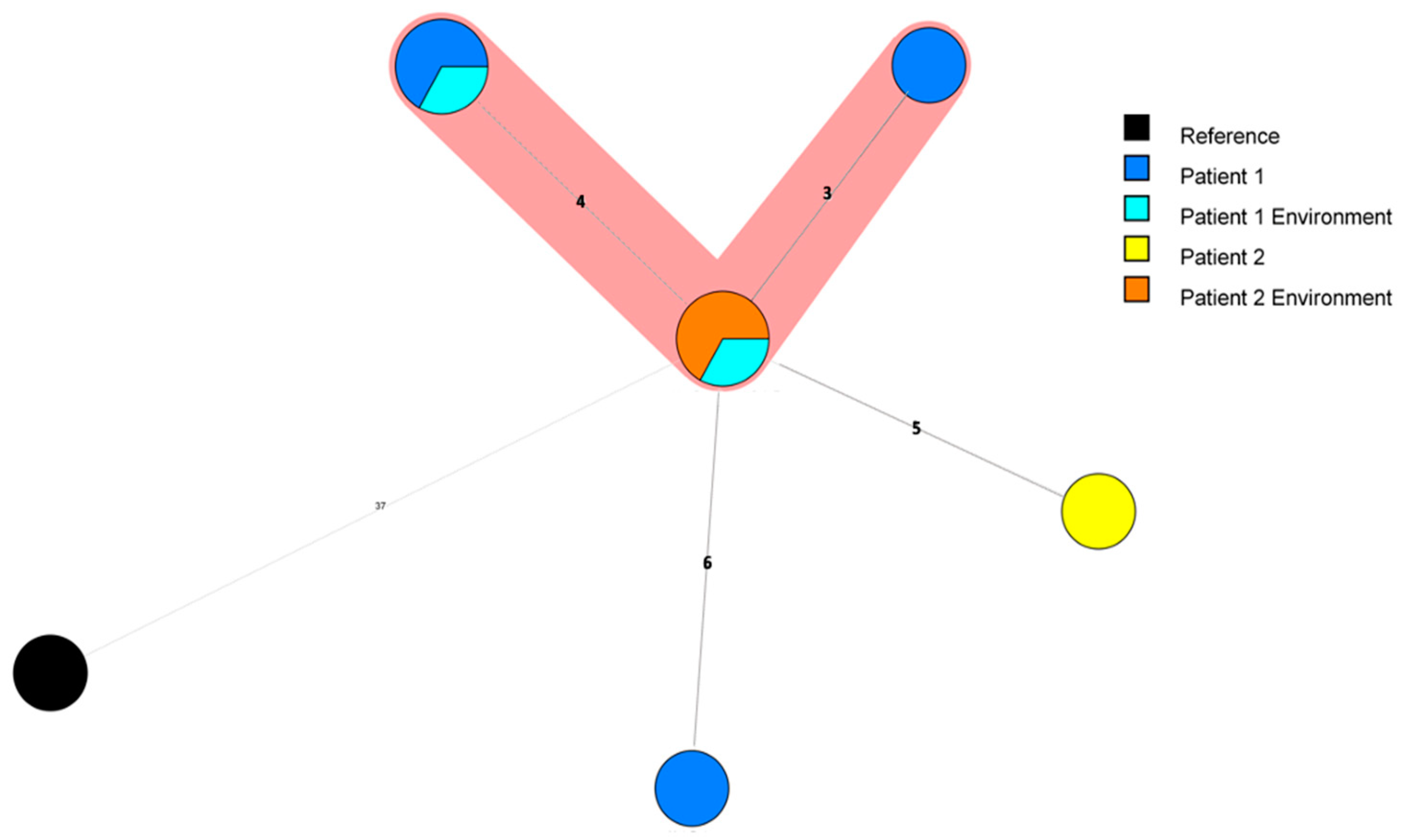
1.2.1. cgMLST of a Panel of 77 ST1 Isolates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis Setting | Reference | Year | Number of Isolates Compared | ST1 Included? | Cluster-Defining Retained Ratio (Maximum SNP or AD) | Number of Alleles Compared | Purpose of Study |
|---|---|---|---|---|---|---|---|
| cgMLST | Moran-Gilad et al. [12] | 2015 | 15 | Yes | 4 AD | 1521 | NA (*) |
| Petzold et al. [17] | 2017 | 45 | No | 4 AD ($) | 1521 | Outbreak | |
| Wüthrich et al. [1] | 2019 | 94 | No | 10 AD | 1521 | Outbreak | |
| Gorzynski et al. [9] | 2022 | 3397 | Yes | 115 AD | 1469 | Epidemiology | |
| Ricci et al. [18] | 2023 | 63 | No | 4 AD ($) | 1346 (+) | Outbreak | |
| wgSNP | Lapierre et al. [21] | 2017 | 77 | Yes | 4 SNP ($) | NA | Outbreak |
| David et al. [8] | 2017 | 229 | Yes | 4 SNP | NA | Outbreak | |
| Schjørring et al. [22] | 2017 | 12 | No | 4 SNP | NA | Outbreak | |
| Wells et al. [23] | 2018 | 28 | Yes | 4 SNP | NA | Epidemiology | |
| Raphael et al. [19] | 2019 | 113 | Yes | no ratio | NA | Epidemiology | |
| cgSNP | Bartley et al. [24] | 2016 | 46 | Yes | no ratio | Not mentioned | Outbreak |
| Qin et al. [25] | 2016 | 53 | Yes | no ratio | 1896 | Epidemiology | |
| Buultjens et al. [26] | 2017 | 180 | No | 9 SNP | NA | Outbreak | |
| Raphael et al. [19] | 2019 | 30 | Yes | 4 SNP ($) | Not mentioned | Outbreak | |
| Gorzynski et al. [9] | 2022 | 3397 | Yes | 16 SNP | 1469 | Epidemiology | |
| Ricci et al. [18] | 2023 | 63 | No | 4 SNP ($) | Not mentioned | Outbreak | |
| wgMLST | Raphael et al. [27] | 2016 | 30 | Yes | >98% similarity | Not mentioned | Outbreak |
| Raphael et al. [19] | 2019 | 113 | Yes | >98% similarity | 5778 | Epidemiology |
1.2.2. wgSNP Analysis of the Panel of 77 ST1 Isolates
1.2.3. wgMLST Analysis of the ST1 Isolates
2. Discussion
3. Conclusions
4. Materials and Methods
4.1. Isolate Preparation, Routine Diagnostics and Typing
4.2. Whole-Genome Sequencing
4.3. Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wüthrich, D.; Gautsch, S.; Spieler-Denz, R.; Dubuis, O.; Gaia, V.; Moran-Gilad, J.; Hinic, V.; Seth-Smith, H.M.; Nickel, C.H.; Tschudin-Sutter, S.; et al. Air-conditioner cooling towers as complex reservoirs and continuous source of Legionella pneumophila infection evidenced by a genomic analysis study in 2017, Switzerland. Eurosurveillance 2019, 24, 1800192. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Harada, S.; Edelstein, P.H.; Nguyen, J.C.; Baddour, L.M.; Edelstein, P.H. Relapsing Legionella pneumophila cellulitis: A case report and review of the literature. J. Infect. Chemother. 2010, 16, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, T.; Schmidt, E.; Papanicolaou, G.; Hauser, J.; Lezcano, C.; Tang, Y.W.; Markova, A. Cutaneous Legionella infections in allogeneic hematopoietic cell transplantation recipients. Dermatol. Online J. 2020, 26, 13030/qt05f926n7. [Google Scholar] [CrossRef]
- Ginevra, C.; Chastang, J.; David, S.; Mentasti, M.; Yakunin, E.; Chalker, V.J.; Chalifa-Caspi, V.; Valinsky, L.; Jarraud, S.; Moran-Gilad, J. A real-time PCR for specific detection of the Legionella pneumophila serogroup 1 ST1 complex. Clin. Microbiol. Infect. 2020, 26, 514.e1–514.e6. [Google Scholar] [CrossRef] [PubMed]
- Echahidi, F.M.C. Activity Report from 2011 to 2022 Reference Centre for Legionella pneumophila UZ Brussel—LHUB-ULB [Internet]. National Reference Centre for Legionella pneumophila, Vrije Universiteit Brussel (VUB), Universitair Ziekenhuis Brussel (UZ Brussel), Department of Microbiology and Infection Control, Brussels. Available online: https://www.sciensano.be/sites/default/files/legionella_2011-2022_nrc_rapport_english_final.pdf (accessed on 16 April 2024).
- European Centre for Disease Prevention and Control—ECDC. Legionnaires’ Disease Annual Epidemiological Report for 2021 [Internet]. Stockholm, Sweden; June 2023. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/legionnaires-disease-annual-epidemiological-report-2021.pdf (accessed on 16 April 2024).
- TSO (The Stationery Offce). Part of Williams Lea. Legionnaires’ Disease Technical Guidance Part 2: The Control of Legionella Bacteria in Hot and Cold Water Systems [Internet]. Health and Safety Executive; 2014 First Published, Second Edition. Available online: https://www.hse.gov.uk/pubns/priced/hsg274part2.pdf (accessed on 16 April 2024).
- David, S.; Afshar, B.; Mentasti, M.; Ginevra, C.; Podglajen, I.; Harris, S.R.; Chalker, V.J.; Jarraud, S.; Harrison, T.G.; Parkhill, J. Seeding and Establishment of Legionella pneumophila in Hospitals: Implications for Genomic Investigations of Nosocomial Legionnaires’ Disease. Clin. Infect. Dis. 2017, 64, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Gorzynski, J.; Wee, B.; Llano, M.; Alves, J.; Cameron, R.; McMenamin, J.; Smith, A.; Lindsay, D.; Fitzgerald, J.R. Epidemiological analysis of Legionnaires’ disease in Scotland: A genomic study. Lancet Microbe 2022, 3, e835–e845. [Google Scholar] [CrossRef] [PubMed]
- Vekens, E.; Soetens, O.; De Mendonça, R.; Echahidi, F.; Roisin, S.; Deplano, A.; Eeckhout, L.; Achtergael, W.; Piérard, D.; Denis, O.; et al. Sequence-based typing of Legionella pneumophila serogroup 1 clinical isolates from Belgium between 2000 and 2010. Eurosurveillance 2012, 17, 20302. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, C.; Rosenwinkel, K.H.; Exner, M.; Verstraete, W.; Suchenwirth, R.; Hartemann, P.; Nogueira, R. Legionella occurrence in municipal and industrial wastewater treatment plants and risks of reclaimed wastewater reuse: Review. Water Res. 2019, 149, 21–34. [Google Scholar] [CrossRef]
- Moran-Gilad, J.; Prior, K.; Yakunin, E.; Harrison, T.G.; Underwood, A.; Lazarovitch, T.; Valinsky, L.; Lück, C.; Krux, F.; Agmon, V.; et al. Design and application of a core genome multilocus sequence typing scheme for investigation of Legionnaires’ disease incidents. Eurosurveillance 2015, 20, 21186. Available online: https://www.eurosurveillance.org/content/10.2807/1560-7917.ES2015.20.28.21186 (accessed on 15 November 2022). [CrossRef] [PubMed]
- Wee, B.A.; Alves, J.; Lindsay, D.S.J.; Klatt, A.B.; Sargison, F.A.; Cameron, R.L.; Pickering, A.; Gorzynski, J.; Corander, J.; Marttinen, P.; et al. Population analysis of Legionella pneumophila reveals a basis for resistance to complement-mediated killing. Nat. Commun. 2021, 12, 7165. [Google Scholar] [CrossRef] [PubMed]
- Kozak-Muiznieks, N.A.; Morrison, S.S.; Mercante, J.W.; Ishaq, M.K.; Johnson, T.; Caravas, J.; Lucas, C.E.; Brown, E.; Raphael, B.H.; Winchell, J.M. Comparative genome analysis reveals a complex population structure of Legionella pneumophila subspecies. Infect. Genet. Evol. 2018, 59, 172–185. [Google Scholar] [CrossRef]
- Haas, W.; Lapierre, P.; Musser, K.A. A Bioinformatic Pipeline for Improved Genome Analysis and Clustering of Isolates during Outbreaks of Legionnaires’ Disease. J. Clin. Microbiol. 2021, 59, e00967-20. [Google Scholar] [CrossRef] [PubMed]
- Krøvel, A.V.; Bernhoff, E.; Austerheim, E.; Soma, M.A.; Romstad, M.R.; Löhr, I.H. Legionella pneumophila in Municipal Shower Systems in Stavanger, Norway; A Longitudinal Surveillance Study Using Whole Genome Sequencing in Risk Management. Microorganisms 2022, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Petzold, M.; Prior, K.; Moran-Gilad, J.; Harmsen, D.; Lück, C. Epidemiological information is key when interpreting whole genome sequence data—Lessons learned from a large Legionella pneumophila outbreak in Warstein, Germany. Eurosurveillance 2017, 22, 17–00137. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.L.; Fillo, S.; Ciammaruconi, A.; Lista, F.; Ginevra, C.; Jarraud, S.; Girolamo, A.; Barbanti, F.; Rota, M.C.; Lindsay, D.; et al. Genome analysis of Legionella pneumophila ST23 from various countries reveals highly similar strains. Life Sci. Alliance 2022, 5, e202101117. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.H.; Huynh, T.; Brown, E.; Smith, J.C.; Ruberto, I.; Getsinger, L.; White, S.; Winchell, J.M. Culture of Clinical Specimens Reveals Extensive Diversity of Legionella pneumophila Strains in Arizona. mSphere 2019, 4, e00649-18. [Google Scholar] [CrossRef] [PubMed]
- Struelens, M.J.; Maes, N.; Rost, F.; Deplano, A.; Jacobs, F.; Liesnard, C.; Bornstein, N.; Grimont, F.; Lauwers, S.; McIntyre, M.P.; et al. Genotypic and Phenotypic Methods for the Investigation of a Nosocomial Legionella pneumophila Outbreak and Efficacy of Control Measures. J. Infect. Dis. 1992, 166, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Nazarian, E.; Zhu, Y.; Wroblewski, D.; Saylors, A.; Passaretti, T.; Hughes, S.; Tran, A.; Lin, Y.; Kornblum, J.; et al. Legionnaires’ Disease Outbreak Caused by Endemic Strain of Legionella pneumophila, New York, New York, USA. Emerg. Infect. Dis. 2017, 23, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Schjørring, S.; Stegger, M.; Kjelsø, C.; Lilje, B.; Bangsborg, J.M.; Petersen, R.F.; David, S.; Uldum, S.A., on behalf of the ESCMID Study Group for Legionella Infections (ESGLI). Genomic investigation of a suspected outbreak of Legionella pneumophila ST82 reveals undetected heterogeneity by the present gold-standard methods, Denmark, July to November Eurosurveillance [Internet]. 2017, 22, 30558. Available online: https://www.eurosurveillance.org/content/10.2807/1560-7917.ES.2017.22.25.30558 (accessed on 16 January 2024).
- Wells, M.; Lasek-Nesselquist, E.; Schoonmaker-Bopp, D.; Baker, D.; Thompson, L.; Wroblewski, D.; Nazarian, E.; Lapierre, P.; Musser, K.A. Insights into the long-term persistence of Legionella in facilities from whole-genome sequencing. Infect. Genet. Evol. 2018, 65, 200–209. [Google Scholar] [CrossRef]
- Bartley, P.B.; Ben Zakour, N.L.; Stanton-Cook, M.; Muguli, R.; Prado, L.; Garnys, V.; Taylor, K.; Barnett, T.C.; Pinna, G.; Robson, J.; et al. Hospital-wide Eradication of a Nosocomial Legionella pneumophila Serogroup 1 Outbreak. Clin. Infect. Dis. 2016, 62, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhang, W.; Liu, W.; Zhou, H.; Ren, H.; Shao, Z.; Lan, R.; Xu, J. Population structure and minimum core genome typing of Legionella pneumophila. Sci. Rep. 2016, 6, 21356. [Google Scholar] [CrossRef] [PubMed]
- Buultjens, A.H.; Chua, K.Y.L.; Baines, S.L.; Kwong, J.; Gao, W.; Cutcher, Z.; Adcock, S.; Ballard, S.; Schultz, M.B.; Tomita, T.; et al. A Supervised Statistical Learning Approach for Accurate Legionella pneumophila Source Attribution during Outbreaks. Appl. Environ. Microbiol. 2017, 83, e01482-17. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.H.; Baker, D.J.; Nazarian, E.; Lapierre, P.; Bopp, D.; Kozak-Muiznieks, N.A.; Morrison, S.S.; Lucas, C.E.; Mercante, J.W.; Musser, K.A.; et al. Genomic Resolution of Outbreak-Associated Legionella pneumophila Serogroup 1 Isolates from New York State. Appl. Environ. Microbiol. 2016, 82, 3582–3590. [Google Scholar] [CrossRef] [PubMed]
- Semenza, J.C.; Ko, A.I. Waterborne Diseases That Are Sensitive to Climate Variability and Climate Change. N. Engl. J. Med. 2023, 389, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Cazalet, C.; Rusniok, C.; Brüggemann, H.; Zidane, N.; Magnier, A.; Ma, L.; Tichit, M.; Jarraud, S.; Bouchier, C.; Vandenesch, F.; et al. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 2004, 36, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Underwood, A.P.; Jones, G.; Mentasti, M.; Fry, N.K.; Harrison, T.G. Comparison of the Legionella pneumophila population structure as determined by sequence-based typing and whole genome sequencing. BMC Microbiol. 2013, 13, 302. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Sánchez-Busó, L.; Harris, S.R.; Marttinen, P.; Rusniok, C.; Buchrieser, C.; Harrison, T.G.; Parkhill, J. Dynamics and impact of homologous recombination on the evolution of Legionella pneumophila. PLoS Genet. 2017, 13, e1006855. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Rusniok, C.; Mentasti, M.; Gomez-Valero, L.; Harris, S.R.; Lechat, P.; Lees, J.; Ginevra, C.; Glaser, P.; Ma, L.; et al. Multiple major disease-associated clones of Legionella pneumophila have emerged recently and independently. Genome Res. 2016, 26, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Mentasti, M.; Tewolde, R.; Aslett, M.; Harris, S.R.; Afshar, B.; Underwood, A.; Fry, N.K.; Parkhill, J.; Harrison, T.G. Evaluation of an Optimal Epidemiological Typing Scheme for Legionella pneumophila with Whole-Genome Sequence Data Using Validation Guidelines. J. Clin. Microbiol. 2016, 54, 2135–2148. [Google Scholar] [CrossRef]
- Lau, K.A.; Gonçalves Da Silva, A.; Theis, T.; Gray, J.; Ballard, S.A.; Rawlinson, W.D. Proficiency testing for bacterial whole genome sequencing in assuring the quality of microbiology diagnostics in clinical and public health laboratories. Pathology 2021, 53, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Gaia, V.; Fry, N.K.; Afshar, B.; Lück, P.C.; Meugnier, H.; Etienne, J.; Peduzzi, R.; Harrison, T.G. Consensus Sequence-Based Scheme for Epidemiological Typing of Clinical and Environmental Isolates of Legionella pneumophila. J. Clin. Microbiol. 2005, 43, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, A.; Crombé, F.; Bosmans, P.; Cools, F.; Piérard, D.; Wybo, I. Serratia marcescens outbreak in a neonatal intensive care unit and the potential of whole-genome sequencing. J. Hosp. Infect. 2021, 111, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michel, C.; Echahidi, F.; Place, S.; Filippin, L.; Colombie, V.; Yin, N.; Martiny, D.; Vandenberg, O.; Piérard, D.; Hallin, M. From Investigating a Case of Cellulitis to Exploring Nosocomial Infection Control of ST1 Legionella pneumophila Using Genomic Approaches. Microorganisms 2024, 12, 857. https://doi.org/10.3390/microorganisms12050857
Michel C, Echahidi F, Place S, Filippin L, Colombie V, Yin N, Martiny D, Vandenberg O, Piérard D, Hallin M. From Investigating a Case of Cellulitis to Exploring Nosocomial Infection Control of ST1 Legionella pneumophila Using Genomic Approaches. Microorganisms. 2024; 12(5):857. https://doi.org/10.3390/microorganisms12050857
Chicago/Turabian StyleMichel, Charlotte, Fedoua Echahidi, Sammy Place, Lorenzo Filippin, Vincent Colombie, Nicolas Yin, Delphine Martiny, Olivier Vandenberg, Denis Piérard, and Marie Hallin. 2024. "From Investigating a Case of Cellulitis to Exploring Nosocomial Infection Control of ST1 Legionella pneumophila Using Genomic Approaches" Microorganisms 12, no. 5: 857. https://doi.org/10.3390/microorganisms12050857