
Comparative Metagenomic Analysis of Bacteriophages and Prophages in Gnotobiotic Mouse Models

 and
and 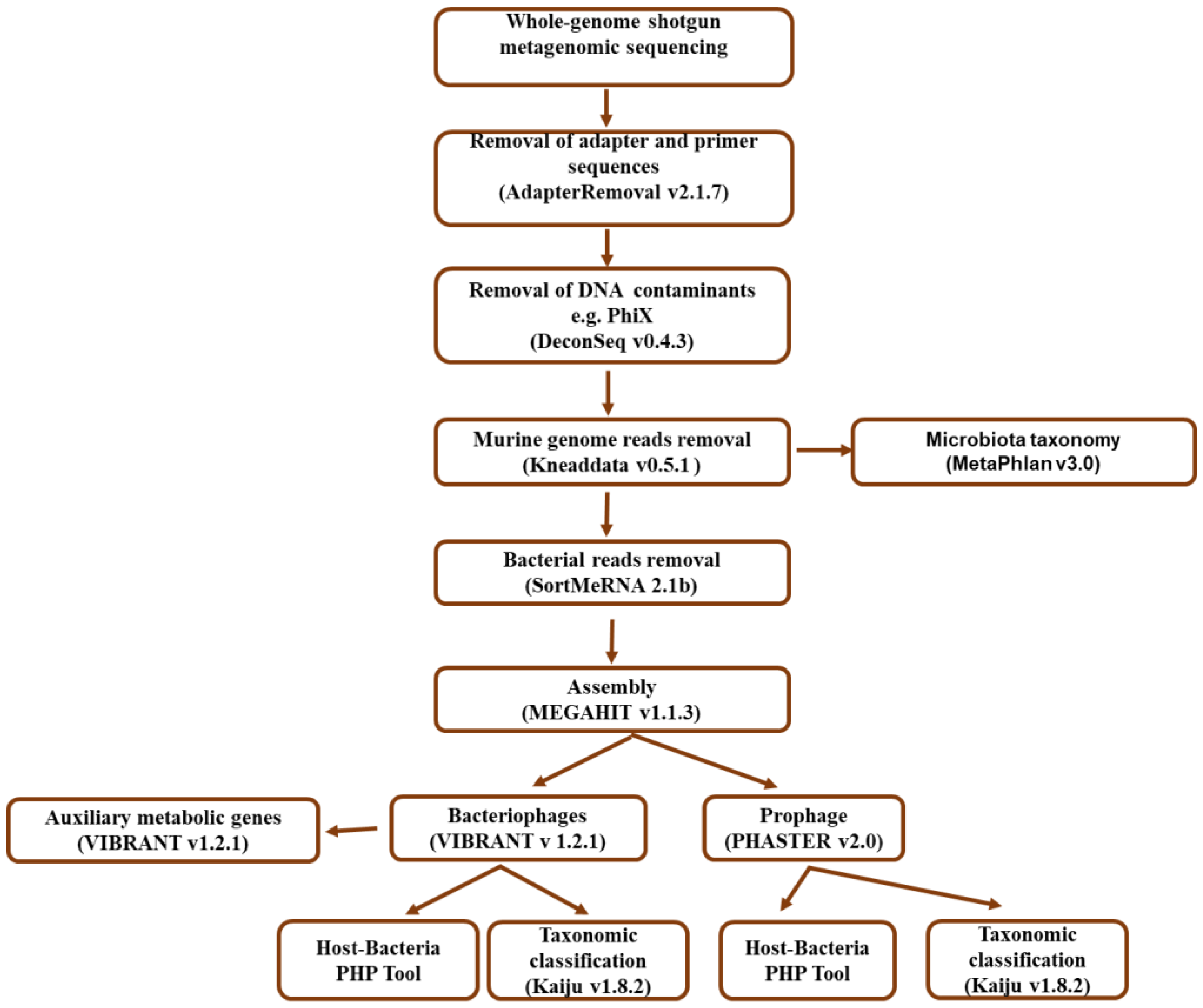{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Models and Sampling
2.2. DNA Extraction
2.3. Metagenomic Library Preparation and Sequencing
2.4. Bioinformatic Analysis Workflow
2.4.1. Quality Filtering and Bacterial Taxonomic Assignment
2.4.2. Bacteriophage Identification and Taxonomic Classification
2.4.3. Prediction and Taxonomic Classification of Prophages
2.5. Statistical Analysis
3. Results
3.1. Sequencing Statistics
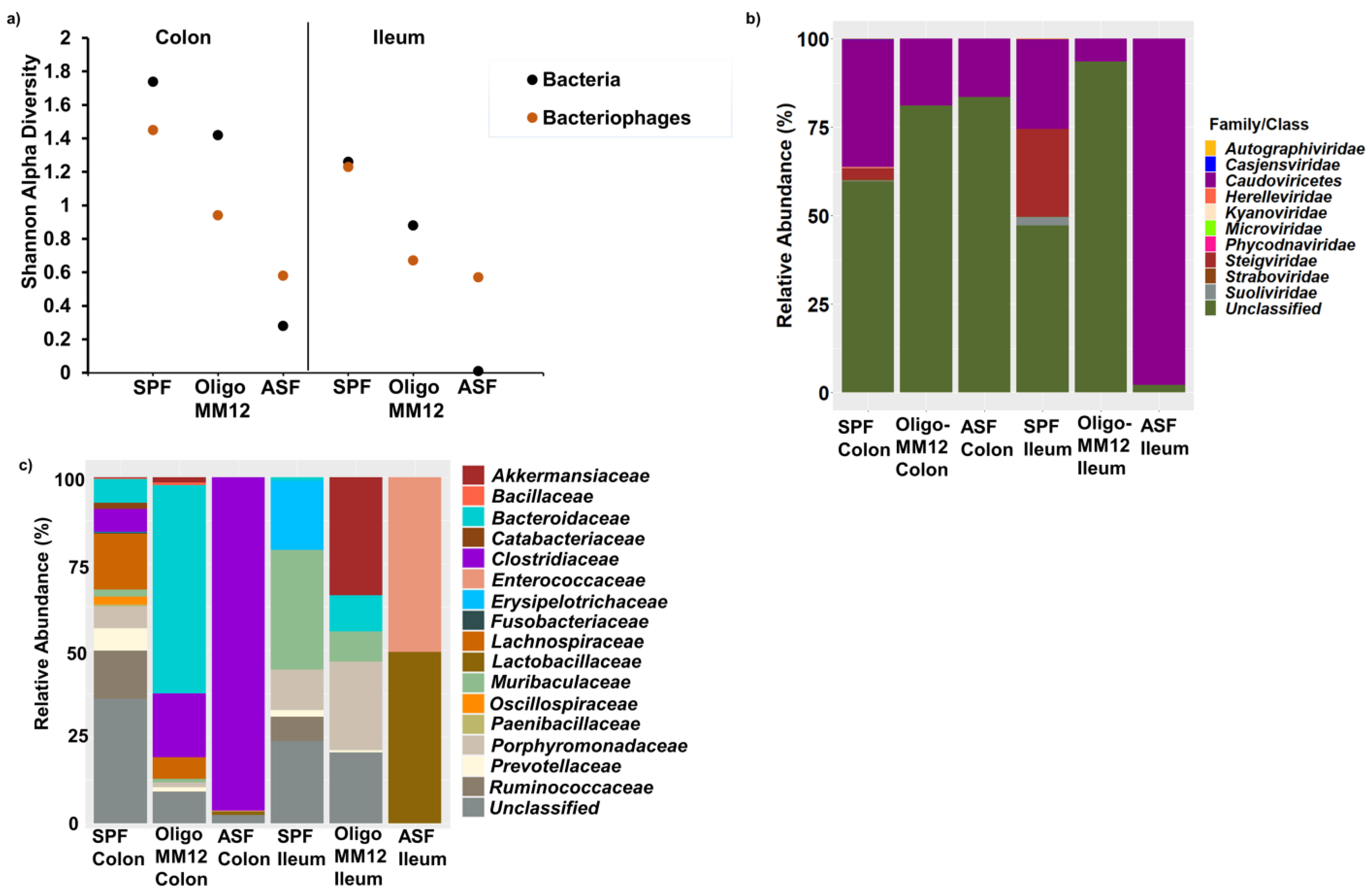
3.2. Bacterial Diversity in the Two Gut Compartments of the Three Murine Models
3.3. Bacteriophages and Predicted Bacterial Hosts
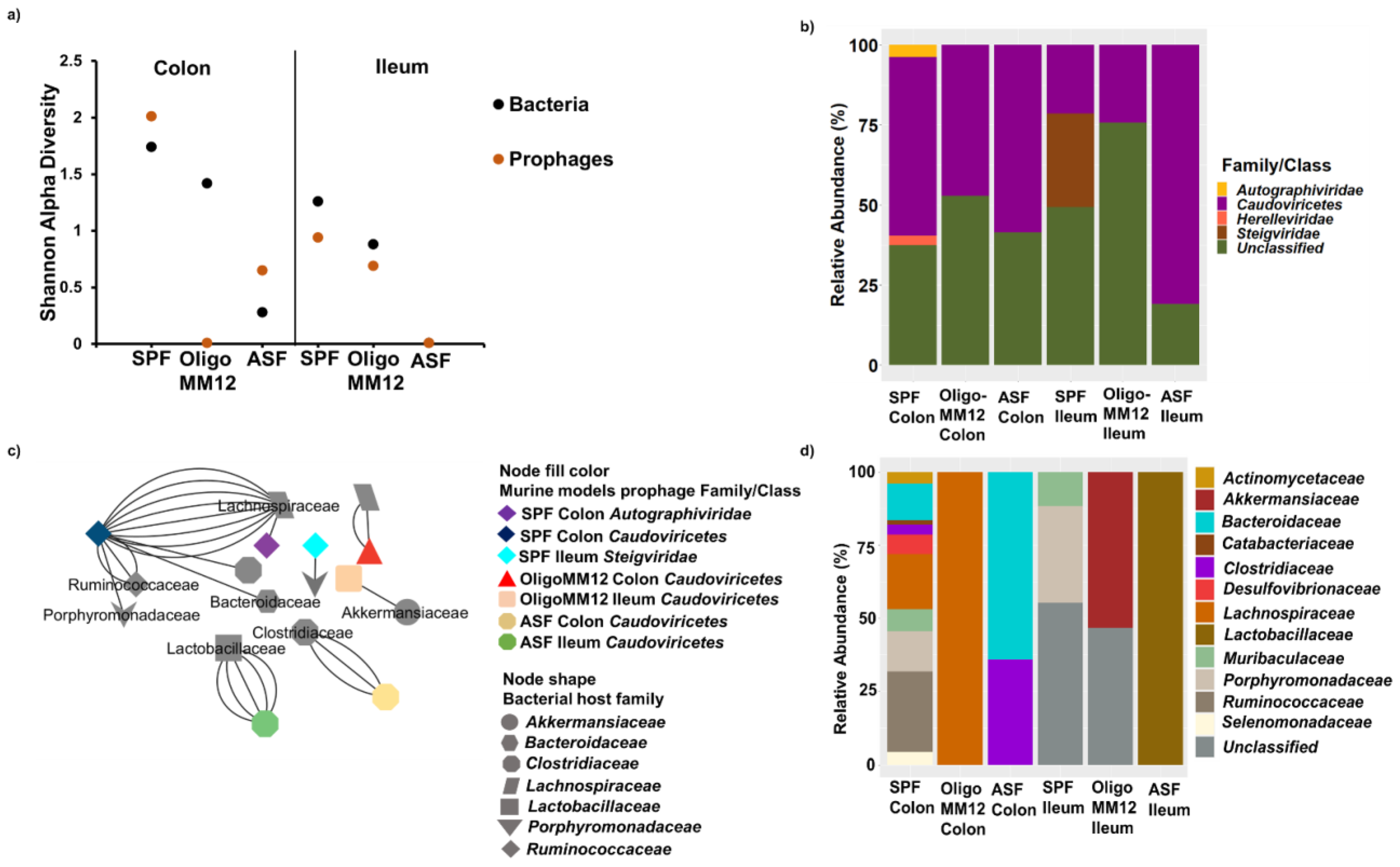
3.4. Taxonomy of Prophages and Their Predicted Bacteria Host
3.5. Auxiliary Metabolic Genes (AMGs) of Bacteriophages
4. Discussion
4.1. Gnotobiotic Oligo-MM12 and ASF Microbiota Consortia Were Detected with Differing Relative Abundances in the Gut
4.2. Bacterial Diversity Triggers Bacteriophage and Prophage Diversity
4.3. AMGs Are Associated with Microbiota Complexity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domínguez-Oliva, A.; Hernández-Ávalos, I.; Martínez-Burnes, J.; Olmos-Hernández, A.; Verduzco-Mendoza, A.; Mota-Rojas, D. The importance of animal models in biomedical research: Current insights and applications. Animals 2023, 13, 1223. [Google Scholar] [CrossRef]
- Choo, J.M.; Rogers, G.B. Establishment of murine gut microbiota in gnotobiotic mice. iScience 2021, 24, 102049. [Google Scholar] [CrossRef]
- Schaedler, R.W.; Dubos, R.; Costello, R. The development of the bacterial flora in the gastrointestinal tract of mice. J. Exp. Med. 1965, 122, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wymore, B.M.; Wannemuehler, M.J.; Phillips, G.J.; Proctor, A.; Overstreet, A.M.; Jergens, A.E.; Orcutt, R.P.; Fox, J.G. The Altered Schaedler Flora: Continued applications of a defined murine microbial community. ILAR J. 2015, 56, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Eberl, C.; Ring, D.; Münch, P.C.; Beutler, M.; Basic, M.; Slack, E.C.; Schwarzer, M.; Srutkova, D.; Lange, A.; Frick, J.S.; et al. Reproducible colonization of germ-free mice with the oligo-mouse-microbiota in different animal facilities. Front. Microbiol. 2020, 10, 2999. [Google Scholar] [CrossRef] [PubMed]
- Garzetti, D.; Brugiroux, S.; Bunk, B.; Pukall, R.; McCoy, K.D.; Macpherson, A.J.; Stecher, B. High-quality whole-genome sequences of the oligo-mouse-microbiota bacterial community. Genome Announc. 2017, 5, e00758-17. [Google Scholar] [CrossRef]
- Brugiroux, S.; Beutler, M.; Pfann, C.; Garzetti, D.; Ruscheweyh, H.J.; Ring, D.; Diehl, M.; Herp, S.; Lötscher, Y.; Hussain, S.; et al. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2016, 2, 21615. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gallego, J.L.; Chou, S.P.; Rienzi, S.C.D.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e5. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.S.; de Vries, L.; Kot, W.; Hansen, L.H.; Castro-Mejia, J.L.; Vogensen, F.K.; Hansen, A.K.; Nielsen, D.S. Mouse Vendor Influence on the Bacterial and Viral Gut Composition Exceeds the Effect of Diet. Viruses 2019, 11, 435. [Google Scholar] [CrossRef]
- Stehr, M.; Greweling, M.C.; Tischer, S.; Singh, M.; Blöcker, H.; Monner, D.A.; Muller, W. Charles River altered Schaedler flora (CRASF®) remained stable for four years in a mouse colony housed in individually ventilated cages. Lab. Anim. 2009, 43, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Lueder, T.; Manefield, M.; Friedrich, M.W. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 2003, 6, 73–78. [Google Scholar] [CrossRef]
- Vestergaard, G.; Schulz, S.; Schöler, A.; Schloter, M. Making big data smarthow to use metagenomics to understand soil quality. Biol. Fertil. Soils 2017, 53, 479–484. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 479–484. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Miguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e17288. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct ide Bruijn/i graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 71, 11257. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, Z.; Cai, Z.; Zhu, Z.; Qiu, Y.; Wu, A.; Jiang, T.; Zheng, H.; Peng, Y. Prokaryotic virus host predictor: A Gaussian model for host prediction of prokaryotic viruses in metagenomics. BMC Biol. 2021, 19, 5. [Google Scholar] [CrossRef]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Poranen, M.M.; et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.; Plichta, D.R.; Nissen, J.N.; Jespersen, M.L.; Shah, S.A.; Deng, L.; Stokholm, J.; Bisgaard, H.; Nielsen, D.S.; Sørensen, S.J.; et al. Genome binning of viral entities from bulk metagenomics data. Nat. Commun. 2022, 13, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Zhou, F.X.; Si, Y.; Yang, H.; Chen, C.; Ren, C.; Wu, J.; Zhang, F. DBSCAN-SWA: An Integrated Tool for Rapid Prophage Detection and Annotation. Front. Genet. 2022, 13, 885048. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, H.; Huang, Y.; Xie, Z.; Zhang, M.; Wei, Y.; Li, J.; Ma, Y.; Luo, M.; Ding, W.; et al. Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep. Genome Biol. 2021, 22, 207. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, 20–25. [Google Scholar] [CrossRef]
- Khan, A.; Wahl, L.M. Quantifying the forces that maintain prophages in bacterial genomes. Theor. Popul. Biol. 2020, 133, 168–179. [Google Scholar] [CrossRef]
- Vazquez-Castellanos, J.F. Diversity Analysis in Viral metagenomes. The Human Virome. Methods Mol. Biol. 2018, 1838, 203–230. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; pp. 275–286. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.5-2. 2018. Available online: https://CRAN.R-project.org/package=vegan (accessed on 12 December 2023).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 131–145. [Google Scholar] [CrossRef]
- Lecuit, M.; Eloit, M. The viruses of the gut microbiota. In The Microbiota in Gastrointestinal Pathophysiology; Academic Press: Cambridge, MA, USA, 2017; pp. 179–183. [Google Scholar]
- von Strempel, A.; Weiss, A.S.; Wittmann, J.; Salvado Silva, M.; Wortmann, E.; Clavel, T.; Debarbieux, L.; Kleigrewe, K.; Stecher, B. Bacteriophages targeting protective commensals impair resistance against Salmonella typhimurium infection in gnotobiotic mice. PLoS Pathog. 2023, 19, e1011600. [Google Scholar] [CrossRef]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 2020, 18, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Yu, P.; Ye, M.; Schwarz, C.; Jiang, X.; Alvarez, P.J.J. Enhanced mutualistic symbiosis between soil phages and bacteria with elevated chromium-induced environmental stress. Microbiome 2021, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Reese, A.T.; Cho, E.H.; Klitzman, B.; Nichols, S.P.; Wisniewski, N.A.; Villa, M.M.; Durand, H.K.; Jiang, S.; Midani, F.S.; Nimmagadda, S.N.; et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. Nature 2018, 19, e35987. [Google Scholar] [CrossRef]
- Blackmer-Raynolds, L.D.; Sampson, T.R. The gut-brain axis goes viral. Cell Host Microbe 2022, 30, 283–285. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Cao, M.M.; Liu, S.Y.; Bi, L.; Chen, S.J.; Wu, H.Y.; Ge, Y.; Han, B.; Zhang, L.; He, J.; Han, L. Distribution Characteristics of Soil Viruses Under Different Precipitation Gradients on the Qinghai-Tibet Plateau. Front. Microbiol. 2022, 13, 848305. [Google Scholar] [CrossRef] [PubMed]
- Kuzyakov, Y.; Mason-Jones, K. Viruses in soil: Nano-scale undead drivers of microbial life, biogeochemical turnover and ecosystem functions. Soil Biol. Biochem. 2018, 1, 305–317. [Google Scholar] [CrossRef]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The gut virome: A new microbiome component in health and disease. EBioMedicine 2022, 81, 104113. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Hedžet, S.; Rupnik, M.; Accetto, T. Novel Siphoviridae bacteriophages infecting Bacteroides uniformis contain diversity generating retroelement. Microorganisms 2021, 9, 892. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, E.; Song, J.; Tong, Y.; Wu, B. Three Salmonella enterica serovar Enteritidis bacteriophages from the Siphoviridae family are promising candidates for phage therapy. Can. J. Microbiol. 2018, 64, 865–875. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Mirzaei, M.K.; Maurice, C.F. Ménage à trois in the human gut: Interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017, 15, 397–408. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, Y.; Wommack, K.E.; Wilhelm, S.W.; Debruyn, J.M.; Sherfy, A.C.; Zhuang, J.; Radosevich, M. Lysogenic reproductive strategies of viral communities vary with soil depth and are correlated with bacterial diversity. Soil Biol. Biochem. 2020, 144, 107767. [Google Scholar] [CrossRef]
- Srinivasiah, S.; Lovett, J.; Ghosh, D.; Roy, K.; Fuhrmann, J.J.; Radosevich, M.; Wommack, K.E. Dynamics of autochthonous soil viral communities parallels dynamics of host communities under nutrient stimulation. FEMS Microbiol. Ecol. 2015, 91, 63. [Google Scholar] [CrossRef]
- Lin, D.M.; Lin, H.C. A theoretical model of temperate phages as mediators of gut microbiome dysbiosis. F1000Research 2019, 8, 997. [Google Scholar] [CrossRef]
- Brown, T.L.; Charity, O.J.; Adriaenssens, E.M. Ecological and functional roles of bacteriophages in contrasting environments: Marine, terrestrial and human gut. Curr. Opin. Microbiol. 2022, 70, 102229. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Bae, J.W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 2018, 12, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage Genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef] [PubMed]
- Bailey, Z.M.; Igler, C.; Wendling, C.C. Prophage maintenance is determined by environment-dependent selective sweeps rather than mutational availability. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gao, E.B.; Huang, Y.; Ning, D. Metabolic genes within cyanophage genomes: Implications for diversity and evolution. Genes 2016, 29, 80. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anderson, R.E.; Buchan, A.; Campbell, B.J.; Hallam, S.J.; Hess, M.; Sullivan, M.B.; Walsh, D.A.; Roux, S.; et al. Ecology of inorganic sulfur auxiliary metabolism in widespread bacteriophages. Nat. Commun. 2021, 12, 3503–3510. [Google Scholar] [CrossRef] [PubMed]
- Heyerhoff, B.; Engelen, B.; Bunse, C. Auxiliary Metabolic Gene Functions in Pelagic and Benthic Viruses of the Baltic Sea. Front. Microbiol. 2022, 13, 863620. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Breister, A.M.; Huss, P.; Linz, A.M.; Zanetakos, E.; Zhou, Z.; Rahlff, J.; Esser, S.P.; Probst, A.J.; Raman, S.; et al. Virus-associated organosulfur metabolism in human and environmental systems. Cell Rep. 2021, 36, 109471. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.; Pell, L.G.; Bona, D.; Tsai, A.; Dai, X.X.; Edwards, A.M.; Hendrix, R.W.; Maxwell, K.L.; Davidson, A.L. Tail Tip Proteins Related to Bacteriophage λ gpL Coordinate an Iron-Sulfur Cluster. J. Mol. Biol. 2013, 425, 2450–2462. [Google Scholar] [CrossRef]
- Bick, J.; Dennis, J.J.; Zylstra, G.J.; Nowack, J.; Leustek, T. Identification of a New Class of 5-Adenylylsulfate (APS) Reductases from Sulfate-Assimilating Bacteria. J. Bacteriol. 2000, 18, 135–142. [Google Scholar] [CrossRef]
- Qin, J.; Ji, B.; Ma, Y.; Liu, X.; Wang, T.; Liu, G.; Li, B.; Wang, G.; Gao, P. Diversity and potential function of pig gut DNA viruses. Heliyon 2023, 9, e14020. [Google Scholar] [CrossRef]
- Zhao, J.; Jing, H.; Wang, Z.; Wang, L.; Jian, H.; Zhang, R.; Xiao, X.; Chen, F.; Jiao, N.; Zhang, Y. Novel Viral Communities Potentially Assisting in Carbon, Nitrogen, and Sulfur Metabolism in the Upper Slope Sediments of Mariana Trench. mSystems 2022, 7, e0135821. [Google Scholar] [CrossRef]
- Summer, E.J.; Gonzalez, C.F.; Bomer, M.; Carlile, T.; Embry, A.; Kucherka, A.M.; Lee, J.; Mebane, L.; Morrison, W.C.; Mark, L.; et al. Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J. Bacteriol. 2006, 188, 255–268. [Google Scholar] [CrossRef]
- İlhan, N. Gut Microbiota and Metabolism. Int. J. Med. Biochem. 2018, 1, 115–128. [Google Scholar] [CrossRef]
- Davila, A.M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.H.; Sanz, Y.; Tome, D. Intestinal luminal nitrogen metabolism: Role of the gut microbiota and consequences for the host. Pharmacol. Res. 2013, 68, 95–107. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishola, O.A.; Kublik, S.; Durai Raj, A.C.; Ohnmacht, C.; Schulz, S.; Foesel, B.U.; Schloter, M. Comparative Metagenomic Analysis of Bacteriophages and Prophages in Gnotobiotic Mouse Models. Microorganisms 2024, 12, 255. https://doi.org/10.3390/microorganisms12020255
Ishola OA, Kublik S, Durai Raj AC, Ohnmacht C, Schulz S, Foesel BU, Schloter M. Comparative Metagenomic Analysis of Bacteriophages and Prophages in Gnotobiotic Mouse Models. Microorganisms. 2024; 12(2):255. https://doi.org/10.3390/microorganisms12020255
Chicago/Turabian StyleIshola, Oluwaseun A., Susanne Kublik, Abilash Chakravarthy Durai Raj, Caspar Ohnmacht, Stefanie Schulz, Bärbel U. Foesel, and Michael Schloter. 2024. "Comparative Metagenomic Analysis of Bacteriophages and Prophages in Gnotobiotic Mouse Models" Microorganisms 12, no. 2: 255. https://doi.org/10.3390/microorganisms12020255