
Accelerating Chloroplast Engineering: A New System for Rapid Generation of Marker-Free Transplastomic Lines of Chlamydomonas reinhardtii
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultivation of Algal Lines
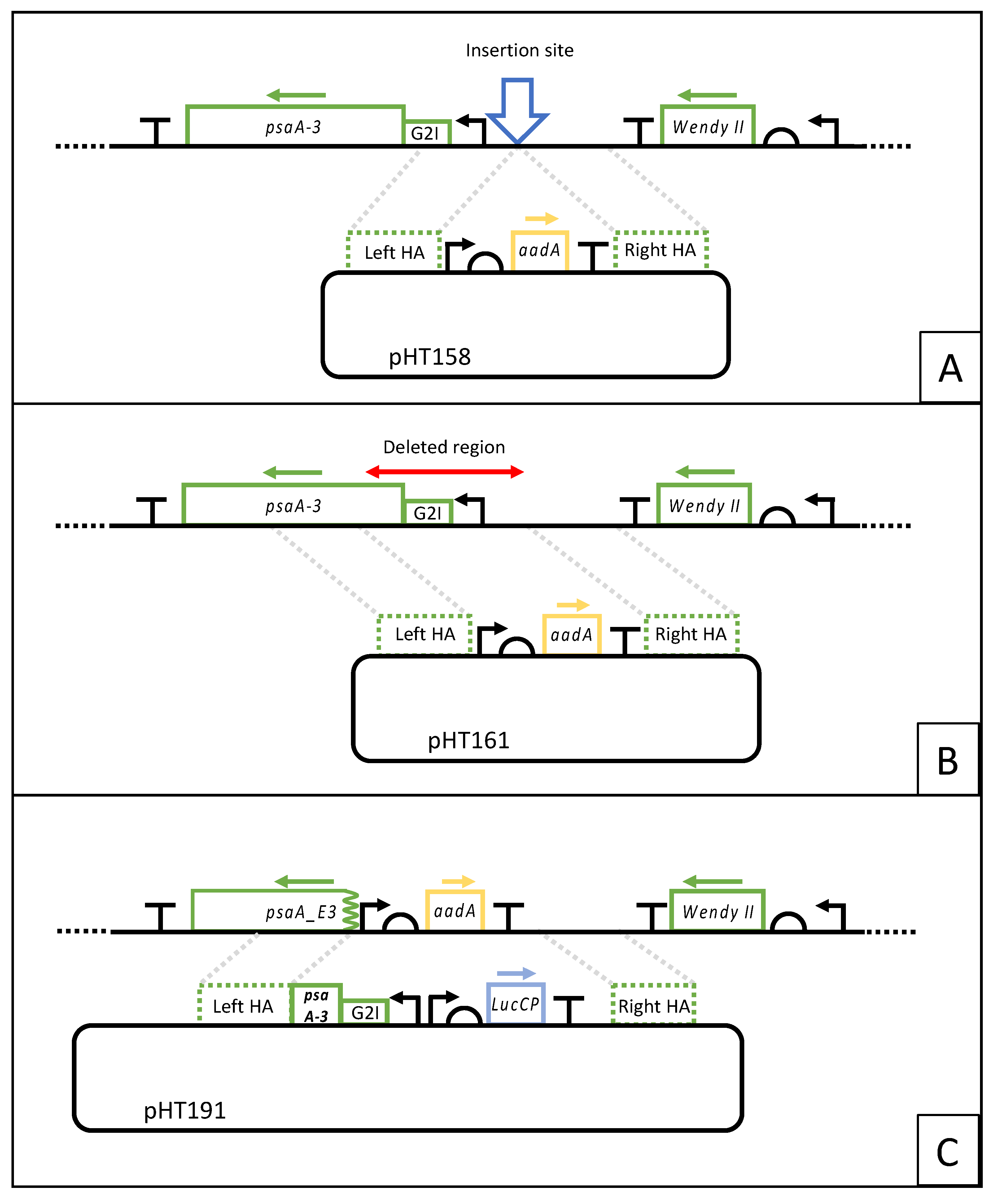
2.2. Construction of Transformation Plasmids
2.3. Transformation of C. reinhardtii
2.4. Genotyping of Transformant Lines
2.5. Luciferase Assays
3. Results
3.1. Creation of a PSI Knockout Mutant and Identification of an Adjacent Neutral Locus for Foreign Gene Insertion
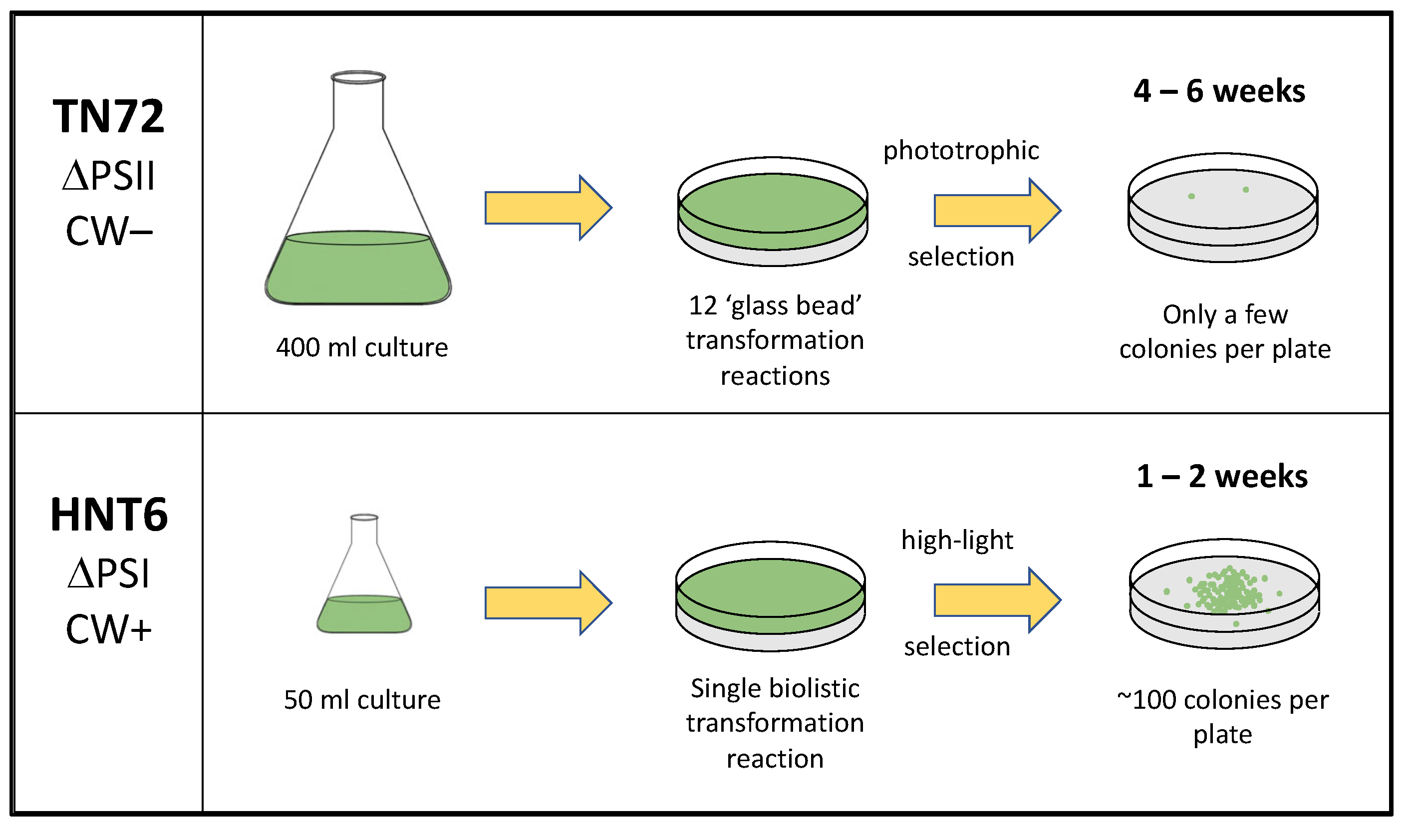
3.2. Strain HNT6 Is Incapable of Phototrophic Growth and Is Sensitive to High Light
3.3. HNT6 Can Serve as a Transformation Recipient Using High Light as the Selection
3.4. Analysis of HNT6 Transformants
3.5. HNT6 Can Be Used for Rapid Parallel Transformation of C. reinhardtii Yielding Marker Free Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Boynton, J.E.; Gillham, N.W.; Harris, E.H.; Hosler, J.P.; Johnson, A.M.; Jones, A.R.; Randolph-Anderson, B.L.; Robertson, D.; Klein, T.M.; Shark, K.B.; et al. Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 1988, 240, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Rochaix, J.-D. Chlamydomonas, a model system for studying the assembly and dynamics of photosynthetic complexes. FEBS Lett. 2002, 529, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimpel, J.A.; Nour-Eldin, H.H.; Scranton, M.A.; Li, D.; Mayfield, S.P. Refactoring the six-gene photosystem II core in the chloroplast of the green algae Chlamydomonas reinhardtii. ACS Synth. Biol. 2016, 5, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.O.; Taunt, H.N.; Mordaka, P.M.; Smith, A.G.; Purton, S. The algal chloroplast as a testbed for synthetic biology designs aimed at radically rewiring plant metabolism. Front. Plant Sci. 2021, 12, 708370. [Google Scholar] [CrossRef]
- Taunt, H.N.; Stoffels, L.; Purton, S. Green biologics: The algal chloroplast as a platform for making biopharmaceuticals. Bioengineered 2018, 9, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Fabris, M.; Abbriano, R.M.; Pernice, M.; Sutherland, D.L.; Commault, A.S.; Hall, C.C.; Labeeuw, L.; McCauley, J.I.; Kuzhiuparambil, U.; Ray, P.; et al. Emerging technologies in algal biotechnology: Toward the establishment of a sustainable, algae-based bioeconomy. Front. Plant Sci. 2020, 11, 279. [Google Scholar] [CrossRef] [Green Version]
- European Food Safety Authority. Opinion of the Scientific Panel on Genetically Modified Organisms on the use of antibiotic resistance genes as marker genes in genetically modified plants. EFSA J. 2004, 48, 1–18. [Google Scholar] [CrossRef]
- Esland, L.; Larrea-Alvarez, M.; Purton, S. Selectable markers and reporter genes for engineering the chloroplast of Chlamydomonas reinhardtii. Biology 2018, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Bendich, A.J. Why do chloroplasts and mitochondria contain so many copies of their genome? BioEssays 1987, 6, 279–282. [Google Scholar] [CrossRef]
- Beacham, T.A.; Sweet, J.B.; Allen, M.J. Large scale cultivation of genetically modified microalgae: A new era for environmental risk assessment. Algal Res. 2017, 25, 90–100. [Google Scholar] [CrossRef]
- Alani, E.; Cao, L.; Kleckner, N. A method for gene disruption that allows repeated use of URA3 Selection in the construction of multiply disrupted yeast strains. Genetics 1987, 116, 541–545. [Google Scholar] [CrossRef]
- Rochaix, J.-D. Chlamydomonas reinhardtii as the photosynthetic yeast. Annu. Rev. Genet. 1995, 29, 209–230. [Google Scholar] [CrossRef]
- Purton, S. Tools and techniques for chloroplast transformation of Chlamydomonas. Adv. Exp. Med. Biol. 2007, 616, 34–45. [Google Scholar] [CrossRef]
- Wannathong, T.; Waterhouse, J.C.; Young, R.E.; Economou, C.K.; Purton, S. New tools for chloroplast genetic engineering allow the synthesis of human growth hormone in the green alga Chlamydomonas reinhardtii. Appl. Microbiol. Biotechnol. 2016, 100, 5467–5477. [Google Scholar] [CrossRef] [Green Version]
- Stoffels, L.; Taunt, H.N.; Charalambous, B.; Purton, S. Synthesis of bacteriophage lytic proteins against Streptococcus pneumoniae in the chloroplast of Chlamydomonas reinhardtii. Plant Biotechnol. J. 2017, 15, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Gangl, D.; Zedler, J.A.Z.; Włodarczyk, A.; Jensen, P.E.; Purton, S.; Robinson, C. Expression and membrane-targeting of an active plant cytochrome P450 in the chloroplast of the green alga Chlamydomonas reinhardtii. Phytochemistry 2015, 110, 22–28. [Google Scholar] [CrossRef]
- Kiataramgul, A.; Maneenin, S.; Purton, S.; Areechon, N.; Hirono, I.; Brocklehurst, T.W.; Unajak, S. An oral delivery system for controlling white spot syndrome virus infection in shrimp using transgenic microalgae. Aquaculture 2020, 521, 735022. [Google Scholar] [CrossRef]
- Charoonnart, P.; Worakajit, N.; Zedler, J.A.Z.; Meetam, M.; Robinson, C.; Saksmerprome, V. Generation of microalga Chlamydomonas reinhardtii expressing shrimp antiviral dsRNA without supplementation of antibiotics. Sci. Rep. 2019, 9, 3164. [Google Scholar] [CrossRef] [Green Version]
- Akram, M.; Khan, M.A.; Ahmed, N.; Bhatti, R.; Pervaiz, R.; Malik, K.; Tahir, S.; Abbas, R.; Ashraf, F.; Ali, Q. Cloning and expression of an anti-cancerous cytokine: Human IL-29 gene in Chlamydomonas reinhardtii. AMB Express 2023, 13, 23. [Google Scholar] [CrossRef]
- Spreitzer, R.J.; Mets, L. Photosynthesis-deficient mutants of Chlamydomonas reinhardii with associated light-sensitive phenotypes. Plant Physiol. 1981, 67, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Erickson, E.; Wakao, S.; Niyogi, K.K. Light stress and photoprotection in Chlamydomonas reinhardtii. Plant J. 2015, 82, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Larrea-Alvarez, M.; Young, R.; Purton, S. A Simple Technology for Generating Marker-Free Chloroplast Transformants of the Green Alga Chlamydomonas reinhardtii. Methods Mol. Biol. 2021, 2317, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, L.; Finlan, A.; Mannall, G.; Purton, S.; Parker, B. Downstream Processing of Chlamydomonas reinhardtii TN72 for Recombinant Protein Recovery. Front. Bioeng. Biotechnol. 2019, 7, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redding, K.; MacMillan, F.; Leibl, W.; Brettel, K.; Hanley, J.; Rutherford, A.W.; Breton, J.; Rochaix, J.D. A systematic survey of conserved histidines in the core subunits of Photosystem I by site-directed mutagenesis reveals the likely axial ligands of P700. EMBO J. 1998, 17, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, E.H. The Chlamydomonas Sourcebook. In A Comprehensive Guide to Biology and Laboratory Use; Academic Press: San Diego, CA, USA, 1989. [Google Scholar] [CrossRef]
- Taylor, G.M.; Mordaka, P.M.; Heap, J.T. Start-Stop Assembly: A functionally scarless DNA assembly system optimized for metabolic engineering. Nucleic Acids Res. 2019, 47, e17. [Google Scholar] [CrossRef] [Green Version]
- Purton, S.; Stevens, D.R.; Muhiuddin, I.P.; Evans, M.C.; Carter, S.; Rigby, S.E.; Heathcote, P. Site-directed mutagenesis of PsaA residue W693 affects phylloquinone binding and function in the photosystem I reaction center of Chlamydomonas reinhardtii. Biochemistry 2001, 40, 2167–2175. [Google Scholar] [CrossRef]
- Kück, U.; Choquet, Y.; Schneider, M.; Dron, M.; Bennoun, P. Structural and transcription analysis of two homologous genes for the P700 chlorophyll a -apoproteins in Chlamydomonas reinhardii: Evidence for in vivo trans-splicing. EMBO J. 1987, 6, 2185–2195. [Google Scholar] [CrossRef]
- Cournac, K.; Redding, K.; Bennoun, P.; Peltier, G. Limited photosynthetic electron flow but no CO2 fixation in Chlamydomonas mutants lacking photosystem I. FEBS Lett. 1997, 416, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt-Clermont, M. Transgenic expression of aminoglycoside adenine transferase in the chloroplast: A selectable marker of site-directed transformation of chlamydomonas. Nucleic Acids Res. 1991, 19, 4083–4089. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Onai, K.; Okamoto, K.; Minagawa, J.; Ishiura, M. Real-time monitoring of chloroplast gene expression by a luciferase reporter: Evidence for nuclear regulation of chloroplast circadian period. Mol. Cell Biol. 2006, 26, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Boynton, J.E.; Gillham, N.W. Chloroplast transformation in Chlamydomonas. Methods Enzymol. 1993, 217, 510–536. [Google Scholar] [CrossRef]
- Lau, K.W.K.; Ren, J.; Wu, M. Redox modulation of chloroplast DNA replication in Chlamydomonas reinhardtii. Antioxid. Redox Signal 2000, 2, 529–535. [Google Scholar] [CrossRef]
- Cavaiuolo, M.; Kuras, R.; Wollman, F.-A.; Choquet, Y.; Vallon, O. Small RNA profiling in Chlamydomonas: Insights into chloroplast RNA metabolism. Nucleic Acids Res. 2017, 45, 10783–10799. [Google Scholar] [CrossRef] [Green Version]
- Blowers, A.D.; Ellmore, G.S.; Klein, U.; Bogorad, L. Transcriptional analysis of endogenous and foreign genes in chloroplast transformants of Chlamydomonas. Plant Cell 1990, 2, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Yoshimura, S.; Ishikura, K.; Takaoka, Y.; Kobayashi, K.; Kato, K.; Shinmyo, A. Effect of coding regions on chloroplast gene expression in Chlamydomonas reinhardtii. J. Biosci. Bioeng. 2003, 95, 276–282. [Google Scholar] [CrossRef]
- Cutolo, E.A.; Mandalà, G.; Dall’Osto, L.; Bassi, R. Harnessing the algal chloroplast for heterologous protein production. Microorganisms 2022, 10, 743. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taunt, H.N.; Jackson, H.O.; Gunnarsson, Í.N.; Pervaiz, R.; Purton, S. Accelerating Chloroplast Engineering: A New System for Rapid Generation of Marker-Free Transplastomic Lines of Chlamydomonas reinhardtii. Microorganisms 2023, 11, 1967. https://doi.org/10.3390/microorganisms11081967
Taunt HN, Jackson HO, Gunnarsson ÍN, Pervaiz R, Purton S. Accelerating Chloroplast Engineering: A New System for Rapid Generation of Marker-Free Transplastomic Lines of Chlamydomonas reinhardtii. Microorganisms. 2023; 11(8):1967. https://doi.org/10.3390/microorganisms11081967
Chicago/Turabian StyleTaunt, Henry N., Harry O. Jackson, Ísarr N. Gunnarsson, Rabbia Pervaiz, and Saul Purton. 2023. "Accelerating Chloroplast Engineering: A New System for Rapid Generation of Marker-Free Transplastomic Lines of Chlamydomonas reinhardtii" Microorganisms 11, no. 8: 1967. https://doi.org/10.3390/microorganisms11081967