

Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture Conditions
2.2. Bacterial Cultures
2.3. Macrophage Infection
2.4. Transfection
2.5. Macrophage Viability
2.6. Reverse Transcriptase-qPCR
2.7. Western Blotting
2.8. Flow Cytometry
2.9. Bacterial Intracellular Survival: Colony-Forming Unit Assay
2.10. Enzymatic Activities of Cathepsins
2.11. Statistical Analysis
3. Results
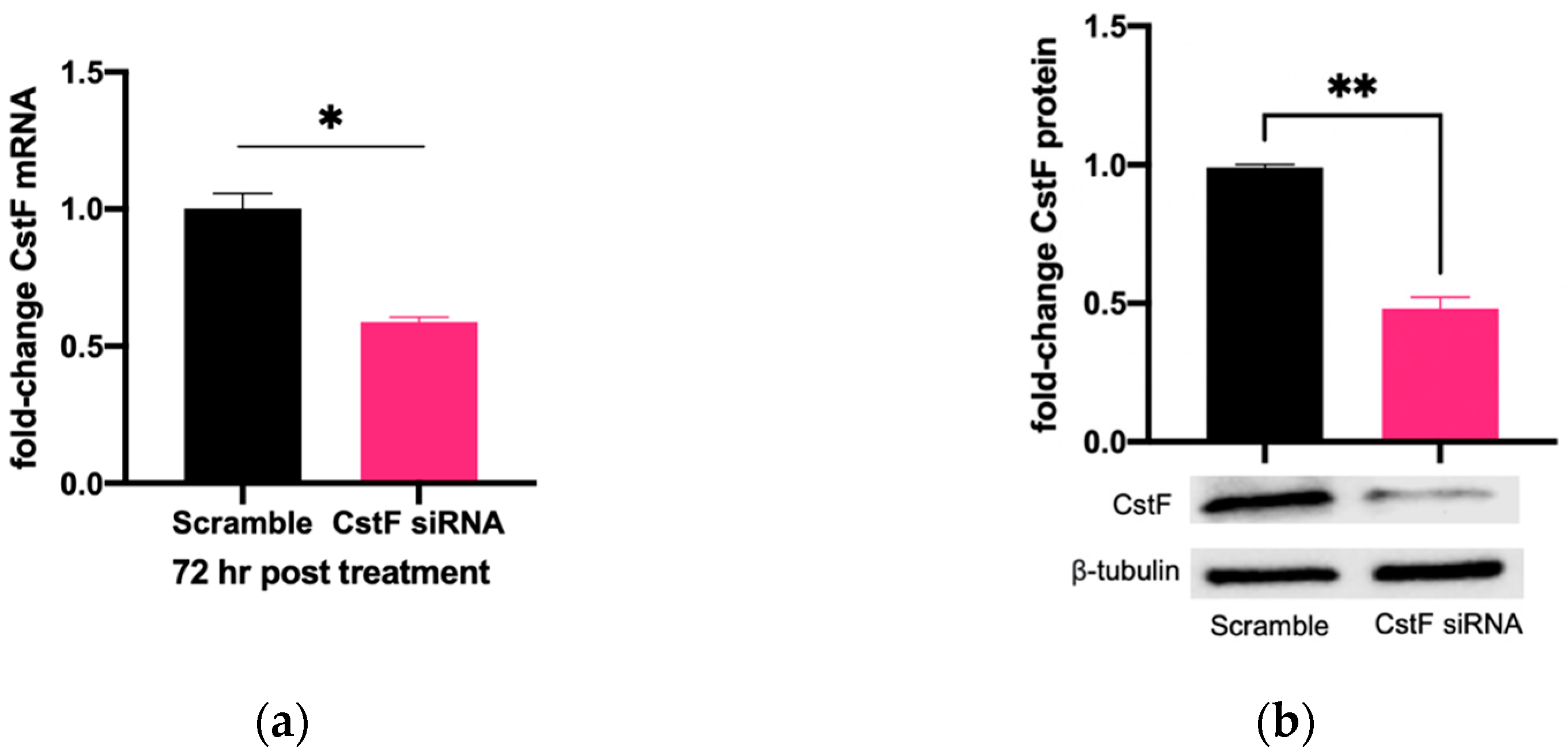
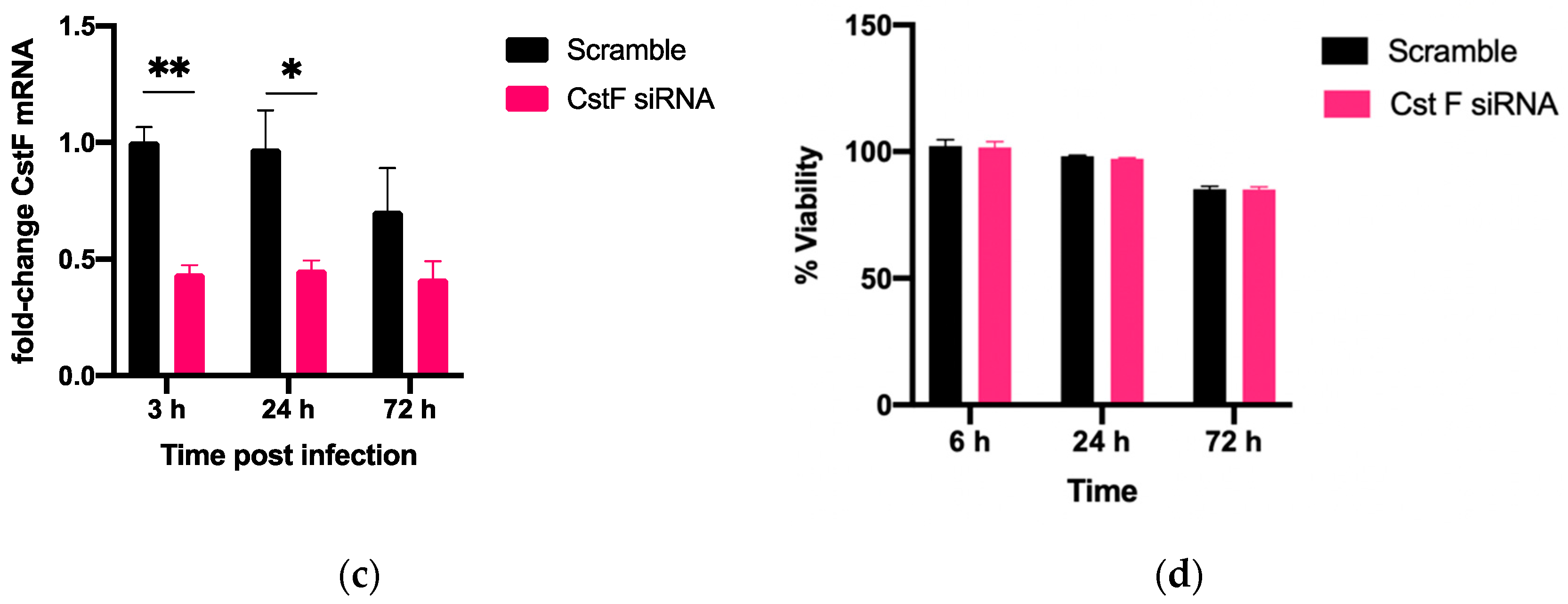
3.1. siRNA-Mediated Gene Silencing Effectively Lowers Cystatin F Expression in Human Primary Macrophages without Cytotoxic Effects
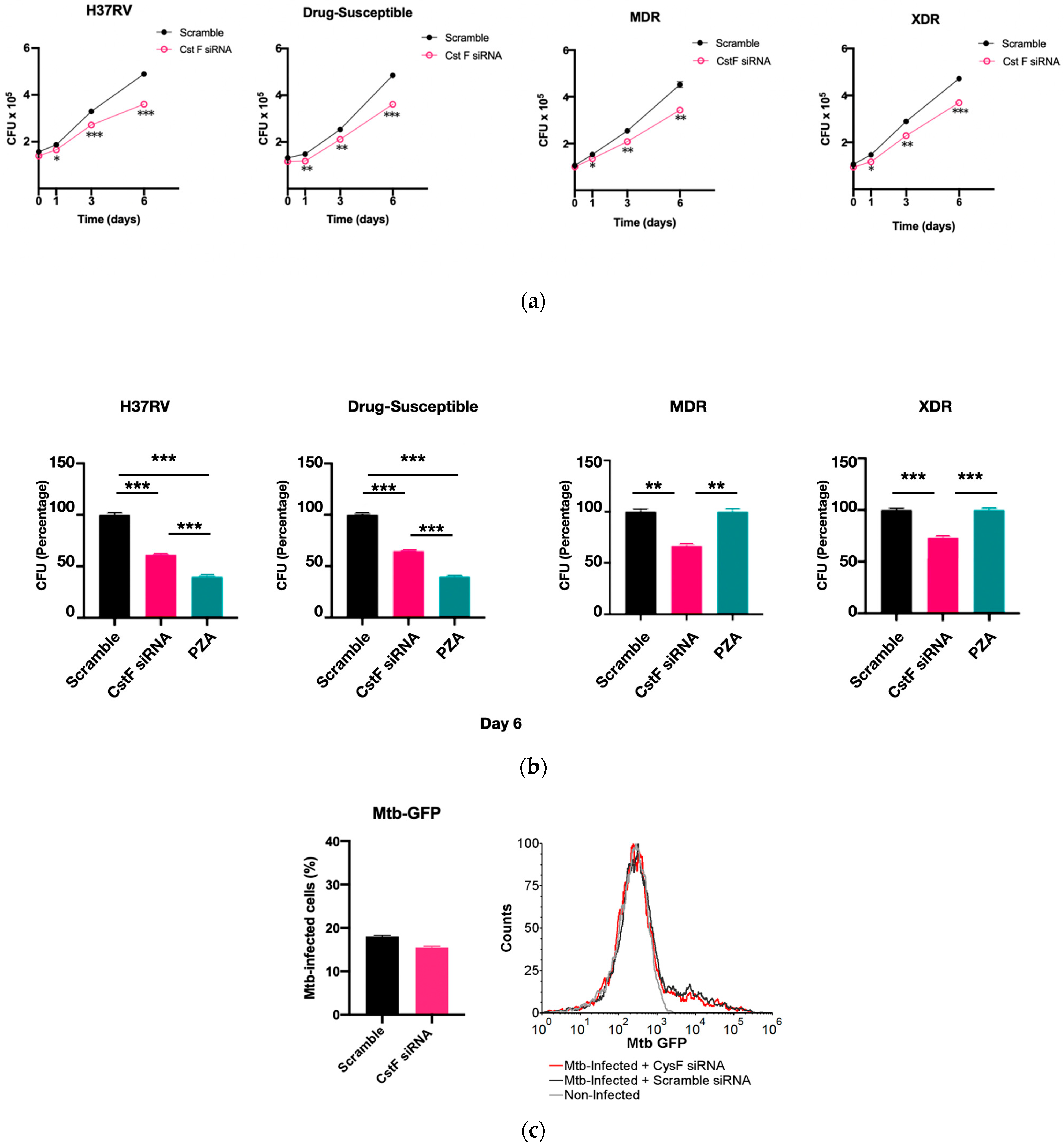
3.2. Silencing of CstF Expression Improves the Intracellular Killing of Mtb Including Clinical Strains with Distinct Drug Resistance Profiles
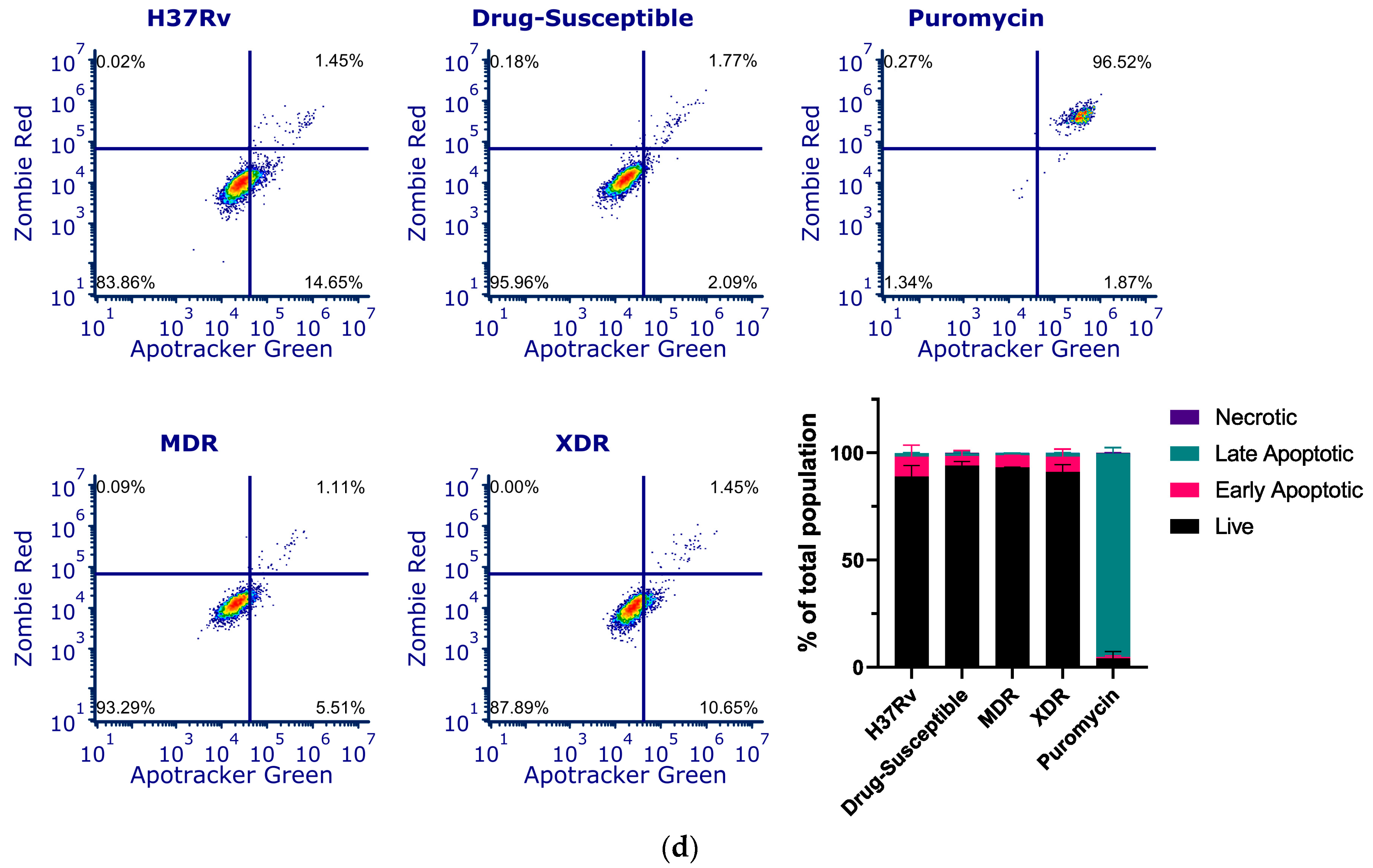
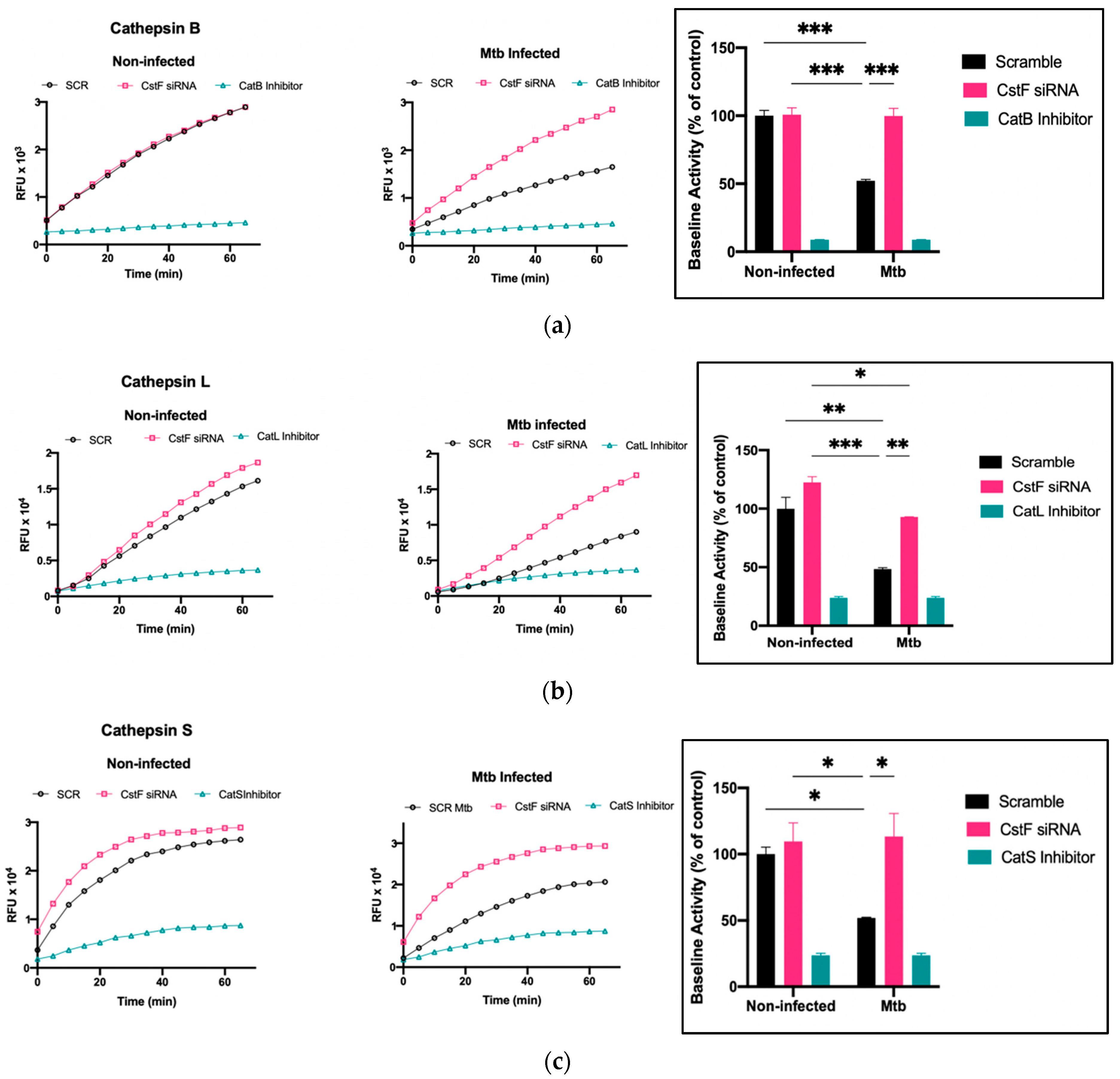
3.3. Silencing of CstF Expression Significantly Impacts Cysteine Cathepsin Enzymatic Activity in Macrophages Infected with Mtb
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022; ISBN 978-92-4-006173-6. [Google Scholar]
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host Evasion and Exploitation Schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef]
- Anes, E.; Azevedo-Pereira, J.M.; Pires, D. Cathepsins and Their Endogenous Inhibitors in Host Defense during Mycobacterium tuberculosis and HIV Infection. Front. Immunol. 2021, 12, 726984. [Google Scholar] [CrossRef] [PubMed]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. ESAT-6 a Major Virulence Factor of Mycobacterium tuberculosis. Biomolecules 2023, 13, 968. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P. Human and Tuberculosis Co-Evolution: An Integrative View. Tuberculosis 2015, 95 (Suppl. S1), S112–S116. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.G. Commentary: Medicine, Population, and Tuberculosis. Int. J. Epidemiol. 2005, 34, 521–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheda, K.; Barry, C.E.; Maartens, G. Tuberculosis. Lancet 2016, 387, 1211–1226. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; McNerney, R.; Murray, M.; Furin, J.; Nardell, E.A.; London, L.; Lessem, E.; et al. The Epidemiology, Pathogenesis, Transmission, Diagnosis, and Management of Multidrug-Resistant, Extensively Drug-Resistant, and Incurable Tuberculosis. Lancet Respir. Med. 2017, 5, 291–360. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Pereira, J.M.; Pires, D.; Calado, M.; Mandal, M.; Santos-Costa, Q.; Anes, E. HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”. Microorganisms 2023, 11, 853. [Google Scholar] [CrossRef]
- Kauffman, K.D.; Sallin, M.A.; Sakai, S.; Kamenyeva, O.; Kabat, J.; Weiner, D.; Sutphin, M.; Schimel, D.; Via, L.; Barry, C.E.; et al. Defective Positioning in Granulomas but Not Lung-Homing Limits CD4 T-Cell Interactions with Mycobacterium tuberculosis-Infected Macrophages in Rhesus Macaques. Mucosal Immunol. 2018, 11, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Ulrichs, T.; Kaufmann, S.H.E. New Insights into the Function of Granulomas in Human Tuberculosis. J. Pathol. 2006, 208, 261–269. [Google Scholar] [CrossRef]
- Peloquin, C.A.; Davies, G.R. The Treatment of Tuberculosis. Clin. Pharmacol. Ther. 2021, 110, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Swaminathan, S. Safety and Tolerability Profile of Second-Line Anti-Tuberculosis Medications. Drug Saf. 2015, 38, 253–269. [Google Scholar] [CrossRef]
- Quenard, F.; Fournier, P.E.; Drancourt, M.; Brouqui, P. Role of Second-Line Injectable Antituberculosis Drugs in the Treatment of MDR/XDR Tuberculosis. Int. J. Antimicrob. Agents 2017, 50, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; Murray, M.; Furin, J.; Nardell, E.A.; Warren, R.M.; Lancet Respiratory Medicine Drug-Resistant Tuberculosis Commission Group. The Lancet Respiratory Medicine Commission: 2019 Update: Epidemiology, Pathogenesis, Transmission, Diagnosis, and Management of Multidrug-Resistant and Incurable Tuberculosis. Lancet Respir. Med. 2019, 7, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Shean, K.; Streicher, E.; Pieterson, E.; Symons, G.; van Zyl Smit, R.; Theron, G.; Lehloenya, R.; Padanilam, X.; Wilcox, P.; Victor, T.C.; et al. Drug-Associated Adverse Events and Their Relationship with Outcomes in Patients Receiving Treatment for Extensively Drug-Resistant Tuberculosis in South Africa. PLoS ONE 2013, 8, e63057. [Google Scholar] [CrossRef]
- Khan, U.; Huerga, H.; Khan, A.J.; Mitnick, C.D.; Hewison, C.; Varaine, F.; Bastard, M.; Rich, M.; Franke, M.F.; Atwood, S.; et al. The EndTB Observational Study Protocol: Treatment of MDR-TB with Bedaquiline or Delamanid Containing Regimens. BMC Infect. Dis. 2019, 19, 733. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Ernst, J.D.; Desvignes, L. Beyond Macrophages: The Diversity of Mononuclear Cells in Tuberculosis. Immunol. Rev. 2014, 262, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G. New Ways to Arrest Phagosome Maturation. Nat. Cell Biol. 2007, 9, 357–359. [Google Scholar] [CrossRef]
- Cambier, C.J.; Takaki, K.K.; Larson, R.P.; Hernandez, R.E.; Tobin, D.M.; Urdahl, K.B.; Cosma, C.L.; Ramakrishnan, L. Mycobacteria Manipulate Macrophage Recruitment through Coordinated Use of Membrane Lipids. Nature 2014, 505, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.K.; Rengarajan, J. Immunology of Mycobacterium tuberculosis Infections. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Refai, A.; Gritli, S.; Barbouche, M.-R.; Essafi, M. Mycobacterium tuberculosis Virulent Factor ESAT-6 Drives Macrophage Differentiation Toward the Pro-Inflammatory M1 Phenotype and Subsequently Switches It to the Anti-Inflammatory M2 Phenotype. Front. Cell. Infect. Microbiol. 2018, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E.M. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530.e3. [Google Scholar] [CrossRef] [Green Version]
- Welin, A.; Eklund, D.; Stendahl, O.; Lerm, M. Human Macrophages Infected with a High Burden of ESAT-6-Expressing M. tuberculosis Undergo Caspase-1- and Cathepsin B-Independent Necrosis. PLoS ONE 2011, 6, e20302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.G.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis Replicates within Necrotic Human Macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. Spatial Localization of Cathepsins: Implications in Immune Activation and Resolution during Infections. Front. Immunol. 2022, 13, 955407. [Google Scholar] [CrossRef]
- Jordao, L.; Bleck, C.K.E.; Mayorga, L.; Griffiths, G.; Anes, E. On the Killing of Mycobacteria by Macrophages. Cell. Microbiol. 2008, 10, 529–548. [Google Scholar] [CrossRef]
- Pires, D.; Marques, J.; Pombo, J.P.; Carmo, N.; Bettencourt, P.; Neyrolles, O.; Lugo-Villarino, G.; Anes, E. Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages. Sci. Rep. 2016, 6, 32247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.; Bernard, E.M.; Pombo, J.P.; Carmo, N.; Fialho, C.; Gutierrez, M.G.; Bettencourt, P.; Anes, E. Mycobacterium tuberculosis Modulates MiR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front. Immunol. 2017, 8, 1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.; Calado, M.; Velez, T.; Mandal, M.; Catalão, M.J.; Neyrolles, O.; Lugo-Villarino, G.; Vérollet, C.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin C in Human Macrophages Improves Anti-Mycobacterial Immune Responses to Mycobacterium tuberculosis Infection and Coinfection With HIV. Front. Immunol. 2021, 12, 742822. [Google Scholar] [CrossRef]
- Bettencourt, P.; Pires, D.; Anes, E. Immunomodulating MicroRNAs of Mycobacterial Infections. Tuberculosis 2016, 97, 1–7. [Google Scholar] [CrossRef]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Mandal, M.; Pinho, J.; Catalão, M.J.; Almeida, A.J.; Azevedo-Pereira, J.M.; Gaspar, M.M.; Anes, E. Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches. Int. J. Mol. Sci. 2023, 24, 1142. [Google Scholar] [CrossRef]
- Pires, D.; Mandal, M.; Matos, A.I.; Peres, C.; Catalão, M.J.; Azevedo-Pereira, J.M.; Satchi-Fainaro, R.; Florindo, H.F.; Anes, E. Development of Chitosan Particles Loaded with SiRNA for Cystatin C to Control Intracellular Drug-Resistant Mycobacterium tuberculosis. Antibiotics 2023, 12, 729. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.I.; Suzuki, T.; Nagai, S.; Yamashita, T.; Toyoda, N.; Matsushima, K. Identification of Genes Specifically Expressed in Human Activated and Mature Dendritic Cells through Serial Analysis of Gene Expression. Blood 2000, 96, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, C.-M.; Wassélius, J.; Wallin, H.; Abrahamson, M. Regulated Expression and Intracellular Localization of Cystatin F in Human U937 Cells. Eur. J. Biochem. 2002, 269, 5502–5511. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F Is a Glycosylated Human Low Molecular Weight Cysteine Proteinase Inhibitor. J. Biol. Chem. 1998, 273, 24797–24804. [Google Scholar] [CrossRef] [Green Version]
- Troegeler, A.; Lastrucci, C.; Duval, C.; Tanne, A.; Cougoule, C.; Maridonneau-Parini, I.; Neyrolles, O.; Lugo-Villarino, G. An Efficient SiRNA-Mediated Gene Silencing in Primary Human Monocytes, Dendritic Cells and Macrophages. Immunol. Cell Biol. 2014, 92, 699–708. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Vanderven, B.C.; Glennie, S.; Mwandumba, H.; Heyderman, R.S. The Macrophage Marches on Its Phagosome: Dynamic Assays of Phagosome Function. Nat. Rev. Immunol. 2009, 9, 594–600. [Google Scholar] [CrossRef] [Green Version]
- Welin, A.; Raffetseder, J.; Eklund, D.; Stendahl, O.; Lerm, M. Importance of Phagosomal Functionality for Growth Restriction of Mycobacterium tuberculosis in Primary Human Macrophages. J. Innate Immun. 2011, 3, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Colbert, J.D.; Schuettelkopf, A.W.; Watts, C. Cystatin F Is a Cathepsin C-Directed Protease Inhibitor Regulated by Proteolysis. EMBO J. 2008, 27, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Halfon, S.; Ford, J.; Foster, J.; Dowling, L.; Lucian, L.; Sterling, M.; Xu, Y.; Weiss, M.; Ikeda, M.; Liggett, D.; et al. Leukocystatin, a New Class II Cystatin Expressed Selectively by Hematopoietic Cells. J. Biol. Chem. 1998, 273, 16400–16408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magister, S.; Kos, J. Cystatins in Immune System. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Lautwein, A.; Burster, T.; Lennon-Duménil, A.-M.; Overkleeft, H.S.; Weber, E.; Kalbacher, H.; Driessen, C. Inflammatory Stimuli Recruit Cathepsin Activity to Late Endosomal Compartments in Human Dendritic Cells. Eur. J. Immunol. 2002, 32, 3348–3357. [Google Scholar] [CrossRef]
- Langerholc, T.; Zavasnik-Bergant, V.; Turk, B.; Turk, V.; Abrahamson, M.; Kos, J. Inhibitory Properties of Cystatin F and Its Localization in U937 Promonocyte Cells. FEBS J. 2005, 272, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.; Enghild, J.J. An Unusual Specificity in the Activation of Neutrophil Serine Proteinase Zymogens. Biochemistry 1990, 29, 5304–5308. [Google Scholar] [CrossRef]
- Kos, J.; Nanut, M.P.; Prunk, M.; Sabotič, J.; Dautović, E.; Jewett, A. Cystatin F as a Regulator of Immune Cell Cytotoxicity. Cancer Immunol. Immunother. 2018, 67, 1931–1938. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, M.; Pires, D.; Catalão, M.J.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis. Microorganisms 2023, 11, 1861. https://doi.org/10.3390/microorganisms11071861
Mandal M, Pires D, Catalão MJ, Azevedo-Pereira JM, Anes E. Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis. Microorganisms. 2023; 11(7):1861. https://doi.org/10.3390/microorganisms11071861
Chicago/Turabian StyleMandal, Manoj, David Pires, Maria João Catalão, José Miguel Azevedo-Pereira, and Elsa Anes. 2023. "Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis" Microorganisms 11, no. 7: 1861. https://doi.org/10.3390/microorganisms11071861