
Molecular Characterization of Salmonella Phage Wara Isolated from River Water in Brazil
 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Purification of Salmonella Phages
2.2. Preparation of High Titer Phage Stocks
2.3. Phage Host Range
2.4. DNA Isolation
2.5. Whole-Genome Sequencing by MinION
2.6. Whole-Genome Sequencing by Illumina
2.7. Genome Annotation and Analysis
2.8. Proteome-Based Clustering and Phylogenetic Analysis
2.9. Electron Microscopy
3. Results
3.1. Isolation of Salmonella Phages
3.2. Electron Microscopy of Wara
3.3. Wara Host Range
3.4. Wara Whole-Genome Sequencing and Assembly
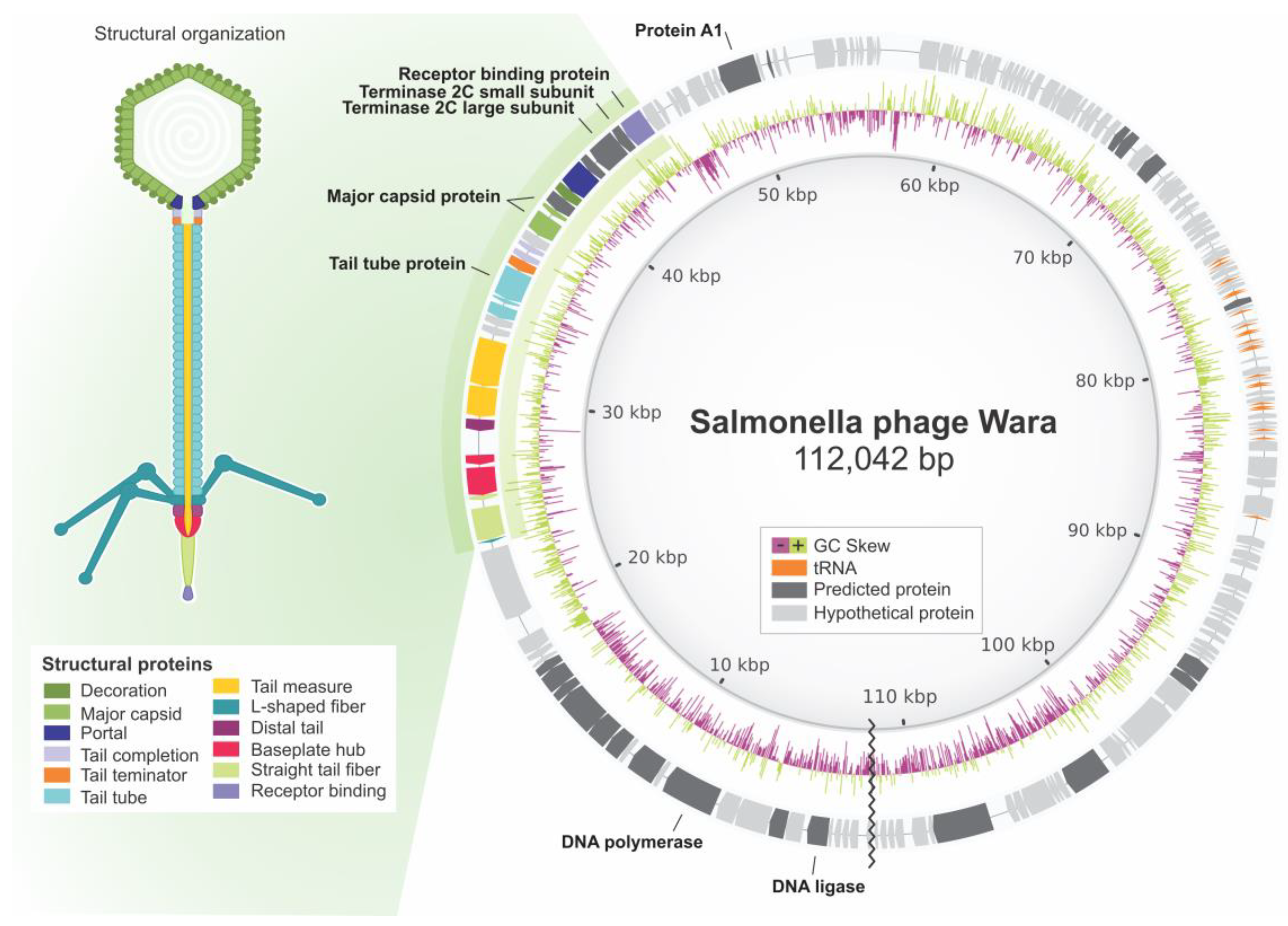
3.5. Wara Genome Annotation and Analysis
3.6. Proteome-Based Clustering and Phylogenetic Analysis of Wara
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Health Topics, Diarrhoea. Available online: https://www.who.int/health-topics/diarrhoea#tab=tab_1 (accessed on 8 May 2023).
- Eng, S.-K.; Pusparajah, P.; Ab Mutalib, N.-S.; Ser, H.-L.; Chan, K.-G.; Lee, L.-H. Salmonella: A Review on Pathogenesis, Epidemiology and Antibiotic Resistance. Front. Life Sci. 2015, 8, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Tondo, E.; Ritter, A. Salmonella and Salmonellosis in Southern Brazil: A Review of the Last Decade. In Salmonella: Classification, Genetics and Disease Outbreaks; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; pp. 175–192. [Google Scholar]
- World Health Statistics. Available online: https://www.who.int/data/gho/publications/world-health-statistics (accessed on 8 May 2023).
- Bryce, A.; Hay, A.D.; Lane, I.F.; Thornton, H.V.; Wootton, M.; Costelloe, C. Global Prevalence of Antibiotic Resistance in Paediatric Urinary Tract Infections Caused by Escherichia Coli and Association with Routine Use of Antibiotics in Primary Care: Systematic Review and Meta-Analysis. BMJ 2016, 352, i939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassim, S.A.A.; Limoges, R.G. Natural Solution to Antibiotic Resistance: Bacteriophages ‘The Living Drugs’. World J. Microbiol. Biotechnol. 2014, 30, 2153–2170. [Google Scholar] [CrossRef] [Green Version]
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas Predators: Understanding and Exploiting Phage–Host Interactions. Nat. Rev. Microbiol. 2017, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Romero-Calle, D.; Guimarães Benevides, R.; Góes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.; Adriaenssens, E.M.; Tolstoy, I.; Kropinski, A.M. Phage Annotation Guide: Guidelines for Assembly and High-Quality Annotation. Phage 2021, 2, 170–182. [Google Scholar] [CrossRef]
- Gibb, B.; Hyman, P.; Schneider, C. The Many Applications of Engineered Bacteriophages—An Overview. Pharmaceuticals 2021, 14, 634. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.; Helwett, E.L. Whole-Genome Sequencing in Outbreak Analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef] [Green Version]
- Goodridge, L.D.; Bisha, B. Phage-Based Biocontrol Strategies to Reduce Foodborne Pathogens in Foods. Bacteriophage 2011, 1, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Spricigo, D.A.; Bardina, C.; Cortés, P.; Llagostera, M. Use of a Bacteriophage Cocktail to Control Salmonella in Food and the Food Industry. Int. J. Food Microbiol. 2013, 165, 169–174. [Google Scholar] [CrossRef]
- Albino, L.A.A.; Rostagno, M.H.; Húngaro, H.M.; Mendonça, R.C.S. Isolation, Characterization, and Application of Bacteriophages for Salmonella Spp. Biocontrol in Pigs. Foodborne Pathog. Dis. 2014, 11, 602–609. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, P.; Zhang, H.; Zhou, Y.; Zhang, L.; Wang, R. Bio-Control of Salmonella Enteritidis in Foods Using Bacteriophages. Viruses 2015, 7, 4836–4853. [Google Scholar] [CrossRef]
- Sukumaran, A.T.; Nannapaneni, R.; Kiess, A.; Sharma, C.S. Reduction of Salmonella on Chicken Meat and Chicken Skin by Combined or Sequential Application of Lytic Bacteriophage with Chemical Antimicrobials. Int. J. Food Microbiol. 2015, 207, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Galarce, N.; Escobar, B.; Rojas, V.; Navarro, C.; Turra, G.; Robeson, J.; Borie, C. Application of a Virulent Bacteriophage Cocktail Leads to Reduction of Salmonella enterica Serovar Enteritidis Counts in Processed Meat Products. Biocontrol Sci. Technol. 2016, 26, 462–475. [Google Scholar] [CrossRef]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the Phages: Defeating Bacterial Defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Santos, L.T.S. Caracterização de metais pesados das águas superficiais da bacia do Rio Subaé (Bahia). Geochim. Bras. 2015, 28, 137–148. [Google Scholar] [CrossRef]
- Pirui, M.; Curso, R.R. Los Bacteriófagos: Del Genoma al Metagenoma; FIBA Fundación para Investigaciones Biológicas Aplicadas: Mar del Plata, Argentina, 2022. [Google Scholar]
- Carey-Smith, G.V.; Billington, C.; Cornelius, A.J.; Hudson, J.A.; Heinemann, J.A. Isolation and Characterization of Bacteriophages Infecting Salmonella spp. FEMS Microbiol. Lett. 2006, 258, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Khan Mirzaei, M.; Nilsson, A.S. Isolation of Phages for Phage Therapy: A Comparison of Spot Tests and Efficiency of Plating Analyses for Determination of Host Range and Efficacy. PLoS ONE 2015, 10, e0118557. [Google Scholar] [CrossRef] [Green Version]
- Romero-Calle, D.X.; Pedrosa-Silva, F.; Tomé, L.M.R.; Sousa, T.J.; de Oliveira Santos, L.T.S.; de Carvalho Azevedo, V.A.; Brenig, B.; Benevides, R.G.; Venancio, T.M.; Billington, C.; et al. Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil. Microorganisms 2022, 11, 111. [Google Scholar] [CrossRef]
- Tomé, L.M.R.; da Silva, F.F.; Fonseca, P.L.C.; Mendes-Pereira, T.; Azevedo, V.A.d.C.; Brenig, B.; Badotti, F.; Góes-Neto, A. Hybrid Assembly Improves Genome Quality and Completeness of Trametes Villosa CCMB561 and Reveals a Huge Potential for Lignocellulose Breakdown. J. Fungi 2022, 8, 142. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Yukgehnaish, K.; Rajandas, H.; Parimannan, S.; Manickam, R.; Marimuthu, K.; Petersen, B.; Clokie, M.R.J.; Millard, A.; Sicheritz-Pontén, T. PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach. Viruses 2022, 14, 342. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic Assignment of Uncultivated Prokaryotic Virus Genomes Is Enabled by Gene-Sharing Networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Gencay, Y.E.; Gambino, M.; Prüssing, T.F.; Brøndsted, L. The Genera of Bacteriophages and Their Receptors Are the Major Determinants of Host Range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, Y.; Confalonieri, F.; Ponchon, L.; Lurz, R.; Chami, M.; Flayhan, A.; Renouard, M.; Huet, A.; Decottignies, P.; Davidson, A.; et al. Insights into Bacteriophage T5 Structure from Analysis of Its Morphogenesis Genes and Protein Components. J. Virol. 2014, 88, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, M.; Askora, A.; Barakat, A.B.; Rabie, O.E.-F.; Hassan, S.E. Isolation and Characterization of Polyvalent Bacteriophages Infecting Multi Drug Resistant Salmonella Serovars Isolated from Broilers in Egypt. Int. J. Food Microbiol. 2018, 266, 8–13. [Google Scholar] [CrossRef]
- Bielke, L.; Higgins, S.; Donoghue, A.; Donoghue, D.; Hargis, B.M. Salmonella Host Range of Bacteriophages That Infect Multiple Genera. Poult. Sci. 2007, 86, 2536–2540. [Google Scholar] [CrossRef]
- Torkashvand, N.; Kamyab, H.; Shahverdi, A.R.; Khoshayand, M.R.; Sepehrizadeh, Z. Isolation, Characterization, and Genome Analysis of a Broad Host Range Salmonella Phage VB_SenS_TUMS_E4: A Candidate Bacteriophage for Biocontrol. Vet. Res. Commun. 2023. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [Green Version]
- Llanos, C.D.; Ortega, J.; Bardales, J.A. Complete Genome Sequences of Five Salmonella Enterica Phages, NBSal001, NBSal002, NBSal003, NBSal004, and NBSal005. Microbiol. Resour. Announc. 2020, 9, e00301-20. [Google Scholar] [CrossRef]
- Rivera, D.; Moreno-Switt, A.I.; Denes, T.G.; Hudson, L.K.; Peters, T.L.; Samir, R.; Aziz, R.K.; Noben, J.-P.; Wagemans, J.; Dueñas, F. Novel Salmonella Phage, VB_Sen_STGO-35-1, Characterization and Evaluation in Chicken Meat. Microorganisms 2022, 10, 606. [Google Scholar] [CrossRef]
- Sattar, S.; Ullah, I.; Khanum, S.; Bailie, M.; Shamsi, B.; Ahmed, I.; Abbas Shah, T.; Javed, S.; Ghafoor, A.; Pervaiz, A.; et al. Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae. Viruses 2022, 14, 241. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salmonella enterica Serovar | Strain | EOP (cf. 14028) | Source */Reference |
|---|---|---|---|
| Enteritidis | ATCC 13076 | 0.88 | LCMG/UFMG |
| Enteritidis | SE3 | 1.12 | DBS/UEFS |
| Enteritidis | SE4 | 0.88 | DBS/UEFS |
| Heidelberg | SH1 | - | DBS/UNIVASF |
| Heidelberg | SH10 | - | DBS/UNIVASF |
| Heidelberg | SH2 | - | DBS/UNIVASF |
| Heidelberg | SH3 | - | DBS/UNIVASF |
| Heidelberg | SH4 | - | DBS/UNIVASF |
| Heidelberg | SH5 | - | DBS/UNIVASF |
| Heidelberg | SH6 | - | DBS/UNIVASF |
| Heidelberg | SH7 | - | DBS/UNIVASF |
| Heidelberg | SH8 | - | DBS/UNIVASF |
| Heidelberg | SH9 | - | DBS/UNIVASF |
| Minnesota | SM1 | 1.29 | DBS/UNIVASF |
| Minnesota | SM10 | 0.88 | DBS/UNIVASF |
| Minnesota | SM2 | - | DBS/UNIVASF |
| Minnesota | SM3 | - | DBS/UNIVASF |
| Minnesota | SM4 | - | DBS/UNIVASF |
| Minnesota | SM5 | - | DBS/UNIVASF |
| Minnesota | SM6 | - | DBS/UNIVASF |
| Minnesota | SM7 | - | DBS/UNIVASF |
| Minnesota | SM8 | - | DBS/UNIVASF |
| Minnesota | SM9 | - | DBS/UNIVASF |
| Typhi | I | - | LCMG/UFMG |
| Typhi | Ia | 1.18 | LCMG/UFMG |
| Typhi | II | - | LCMG/UFMG |
| Typhi | III | - | LCMG/UFMG |
| Typhi | IV | - | LCMG/UFMG |
| Typhimurium | 14028 | 1.00 | ATCC |
| Typhimurium | 14088 | 0.88 | ATCC |
| Typhimurium | II | 1.00 | LCMG/UFMG |
| Typhimurium | III | 1.29 | LCMG/UFMG |
| Typhimurium | IV | - | LCMG/UFMG |
| Assembly Method | Racon | SPAdes | Unicycler | |
|---|---|---|---|---|
| Sequencing Platform | MinION | HiSeq | MinION/HiSeq | Reference NC_048009.1 |
| Number of contigs ≥ 0 bp | 1 | 112 | 1 | 1 |
| Number of contigs ≥ 50,000 bp | 1 | 1 | 1 | 1 |
| Largest contigs (bp) | 112,042 | 110,012 | 110,012 | 110,091 |
| Total length ≥ 50,000 bp | 112,042 | 110,012 | 110,012 | 110,091 |
| GC (%) | 39.15 | 39.17 | 39.17 | 39.22 |
| N50 | 112,042 | 110,012 | 110,012 | 110,091 |
| L50 | 1 | 1 | 1 | 1 |
| Feature | Reference NC_048009.1 | Racon/ MinION | SPAdes/ HiSeq | Unicycler/ HiSeq + MinION |
|---|---|---|---|---|
| CDS | 155 | 170 | 161 | 156 |
| tRNA | 23 | 21 | 21 | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Calle, D.X.; Pedrosa-Silva, F.; Ribeiro Tomé, L.M.; Fonseca, V.; Guimarães Benevides, R.; de Oliveira Santos, L.T.S.; de Oliveira, T.; da Costa, M.M.; Alcantara, L.C.J.; de Carvalho Azevedo, V.A.; et al. Molecular Characterization of Salmonella Phage Wara Isolated from River Water in Brazil. Microorganisms 2023, 11, 1837. https://doi.org/10.3390/microorganisms11071837
Romero-Calle DX, Pedrosa-Silva F, Ribeiro Tomé LM, Fonseca V, Guimarães Benevides R, de Oliveira Santos LTS, de Oliveira T, da Costa MM, Alcantara LCJ, de Carvalho Azevedo VA, et al. Molecular Characterization of Salmonella Phage Wara Isolated from River Water in Brazil. Microorganisms. 2023; 11(7):1837. https://doi.org/10.3390/microorganisms11071837
Chicago/Turabian StyleRomero-Calle, Danitza Xiomara, Francisnei Pedrosa-Silva, Luiz Marcelo Ribeiro Tomé, Vagner Fonseca, Raquel Guimarães Benevides, Leila Thaise Santana de Oliveira Santos, Tulio de Oliveira, Mateus Matiuzzi da Costa, Luiz Carlos Junior Alcantara, Vasco Ariston de Carvalho Azevedo, and et al. 2023. "Molecular Characterization of Salmonella Phage Wara Isolated from River Water in Brazil" Microorganisms 11, no. 7: 1837. https://doi.org/10.3390/microorganisms11071837