
The Gut Microbiota Contributes to Systemic Responses and Liver Injury in Gut-Derived Sepsis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
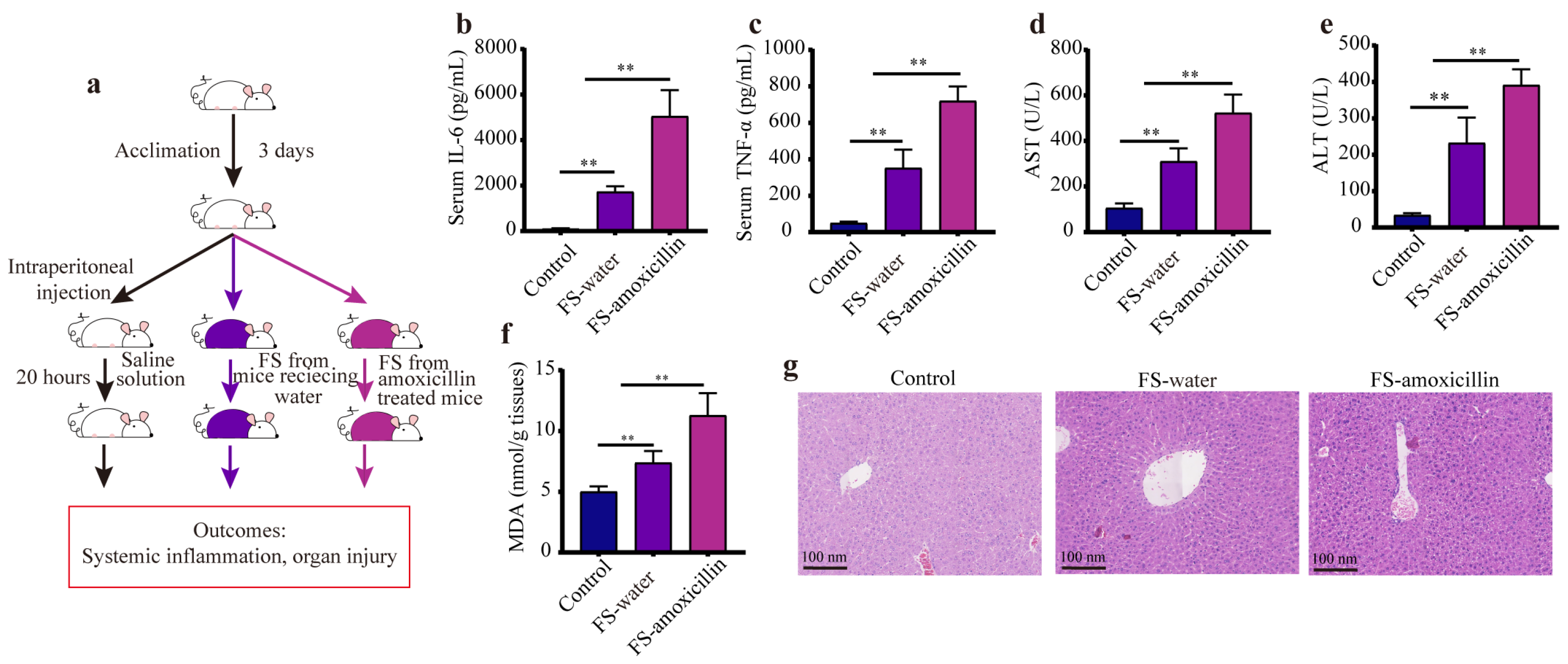
2.2. Fecal Suspension Intraperitoneal Injection Experiments
2.3. RNA Sequencing and RT-qPCR
2.4. 16S rRNA Sequencing
2.5. Biochemistry Experiments
2.6. Statistical Analysis
3. Results
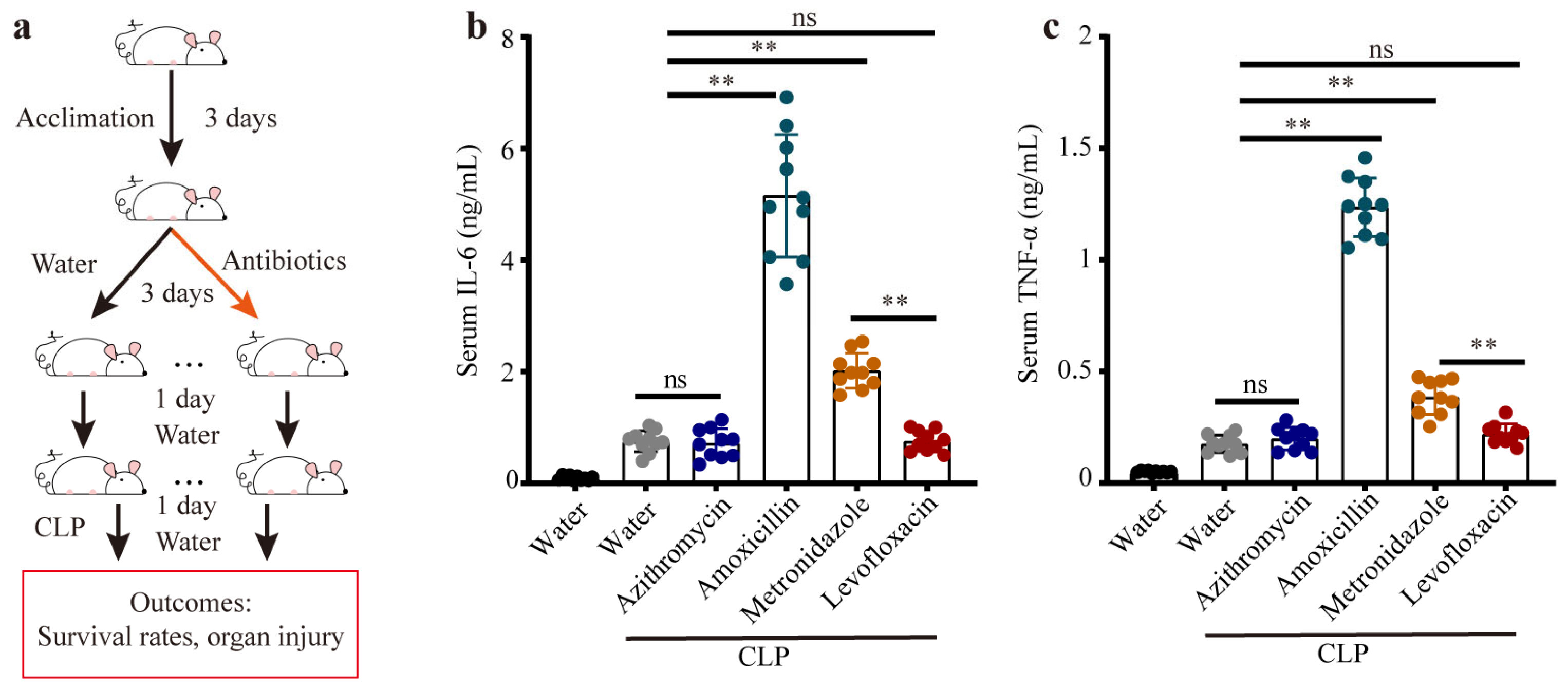
3.1. Distinct Gut Microbiota Influenced the Systemic Injury in Sepsis Mice
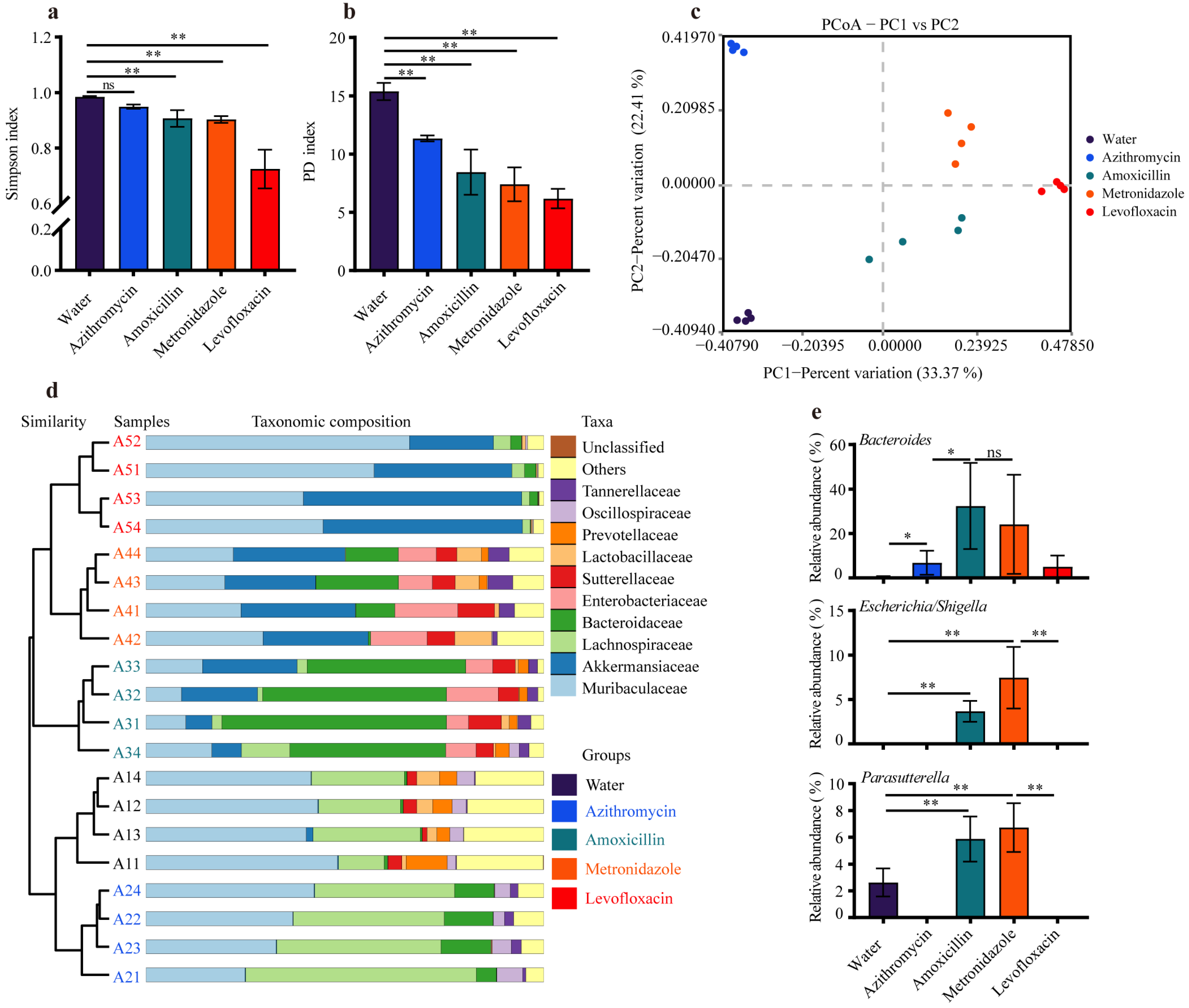
3.2. Previous Antibiotic Exposure Caused Different Enterotypes in Mice
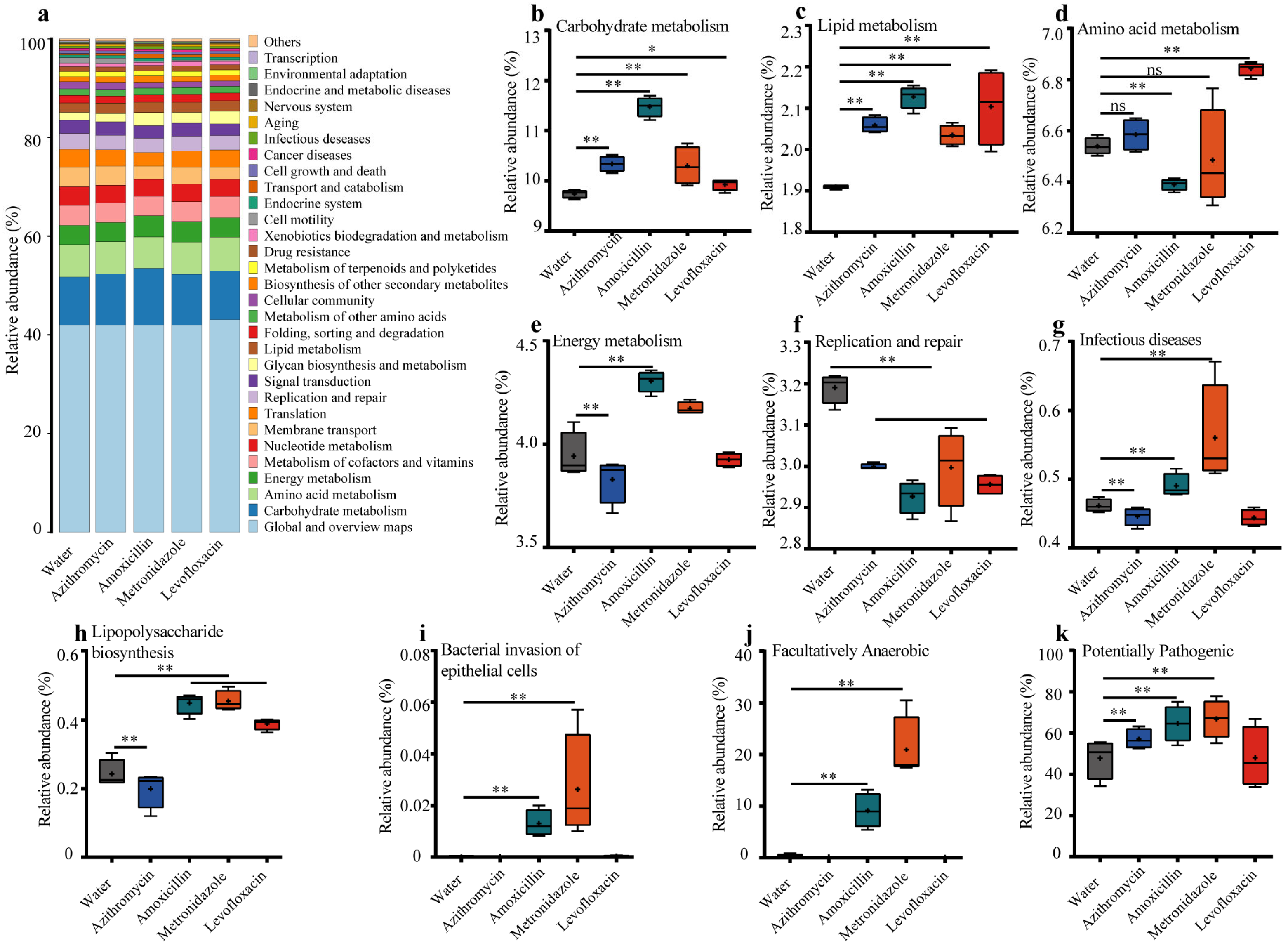
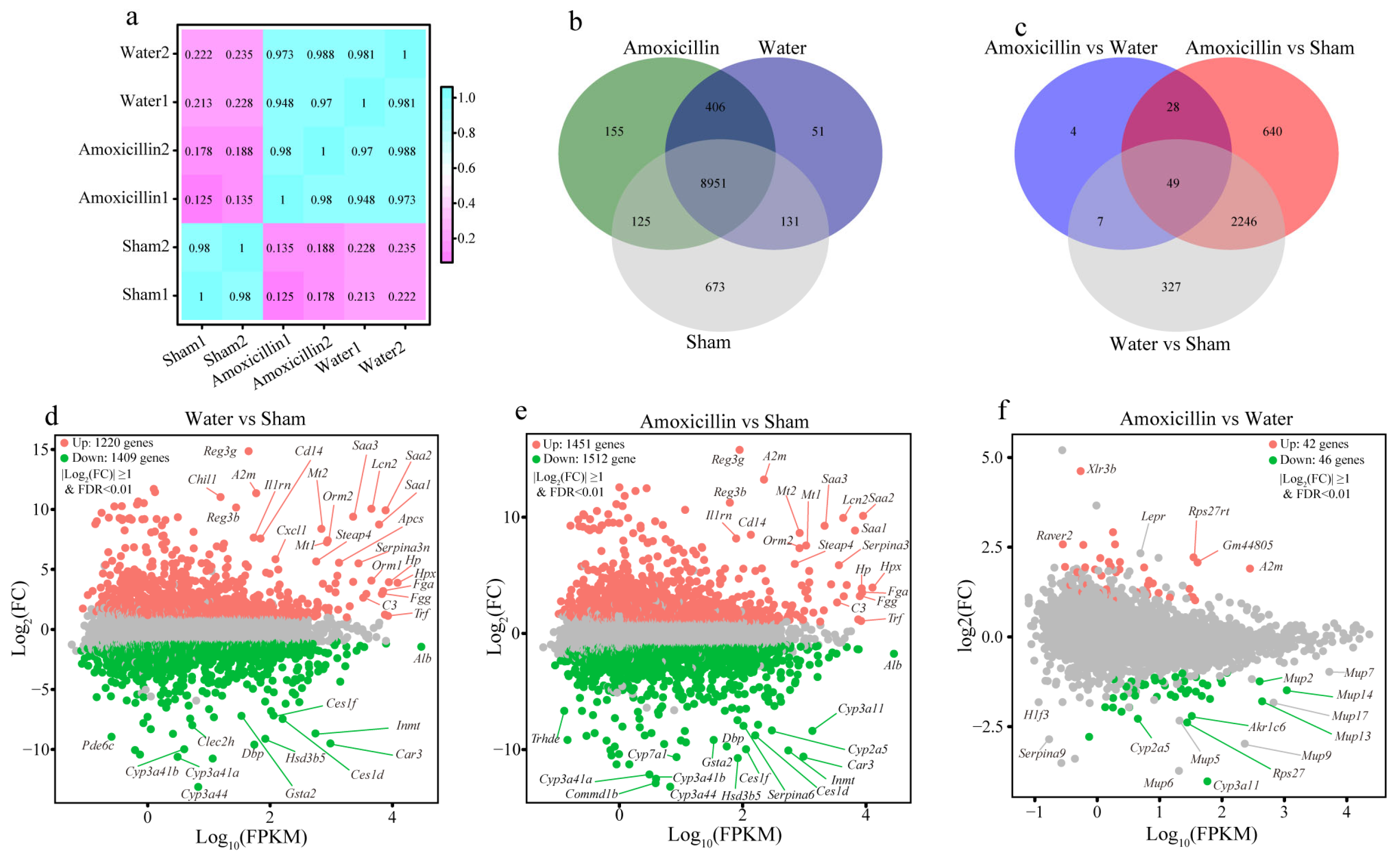
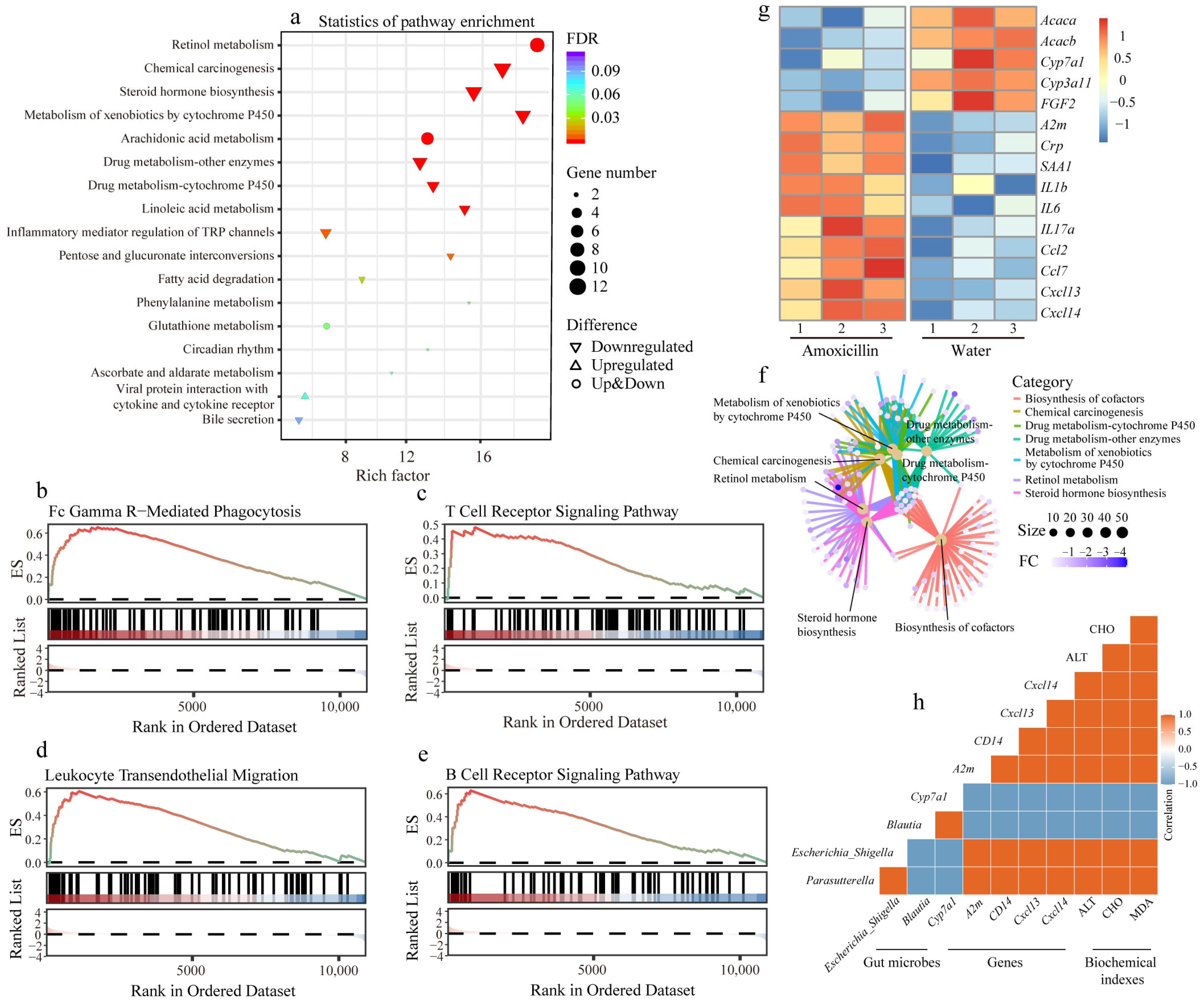
3.3. Previous Antibiotic Exposure Disordered the Function of Gut Microbiota in Mice
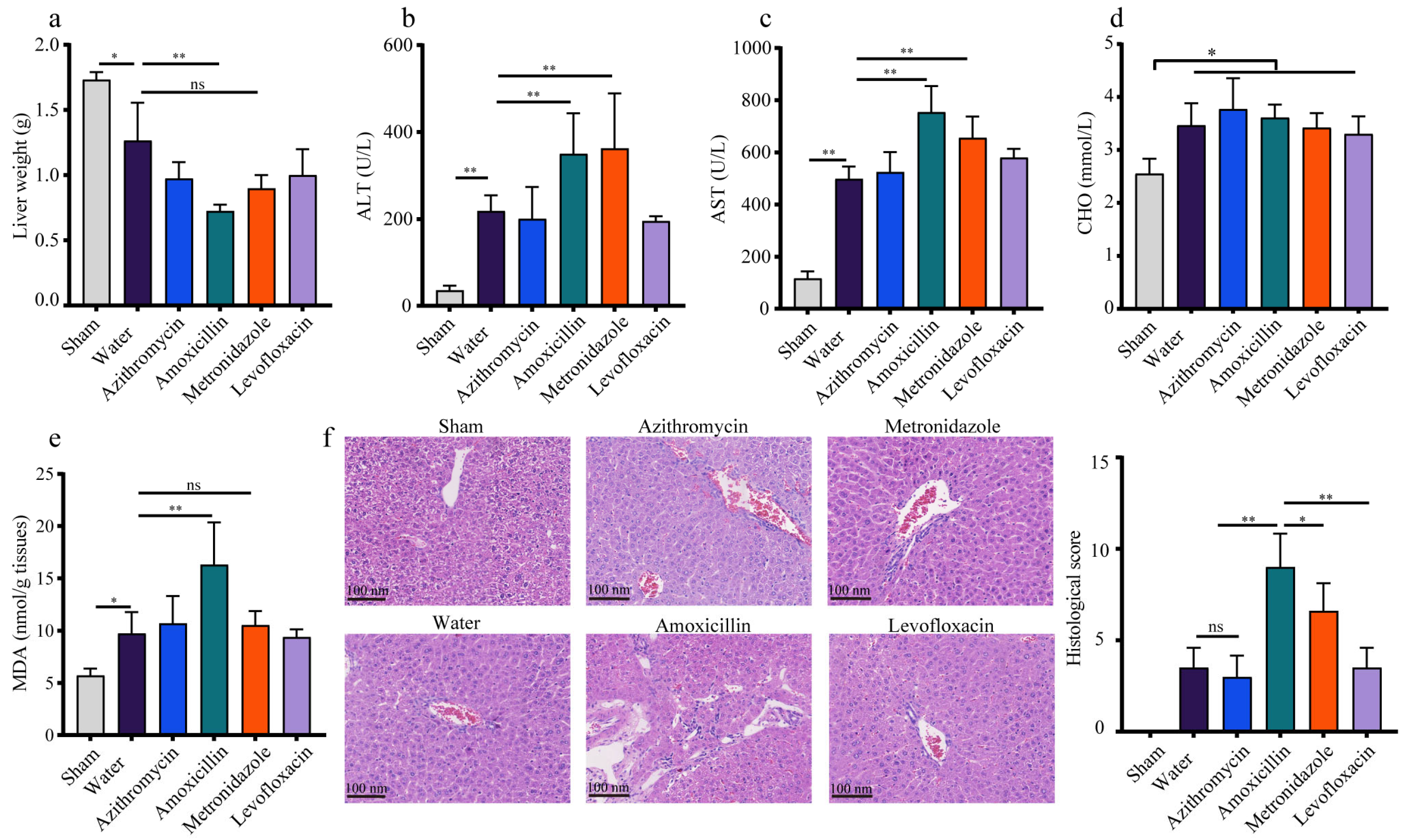
3.4. The Polymicrobial Sepsis Impaired the Liver Function in Mice
3.5. Previous Administration of Amoxicillin Exaggerated the Sepsis-Induced Liver Injury in Mice
3.6. The Enterotypes Enriched in Pathogenic Microorganisms Aggravated Systemic Inflammation and Liver Injury in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- WHO Reports of Sepsis. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/sepsis (accessed on 26 August 2020).
- Fohner, A.E.; Greene, J.D.; Lawson, B.L.; Chen, J.H.; Kipnis, P.; Escobar, G.J.; Liu, V.X. Assessing clinical heterogeneity in sepsis through treatment patterns and machine learning. J. Am. Med. Inform. Assoc. 2019, 26, 1466–1477. [Google Scholar] [CrossRef]
- Leligdowicz, A.; Matthay, M.A. Heterogeneity in sepsis: New biological evidence with clinical applications. Crit. Care 2019, 23, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Wastyk, H.C.; Fragiadakis, G.K.; Perelman, D.; Dahan, D.; Merrill, B.D.; Yu, F.B.; Topf, M.; Gonzalez, C.G.; Van Treuren, W.; Han, S.; et al. Gut-microbiota-targeted diets modulate human immune status. Cell 2021, 184, 4137–4153.e14. [Google Scholar] [CrossRef] [PubMed]
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19. [Google Scholar] [CrossRef]
- Kudelka, M.R.; Stowell, S.R.; Cummings, R.D.; Neish, A.S. Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 597–617. [Google Scholar] [CrossRef]
- Stevens, B.R.; Goel, R.; Seungbum, K.; Richards, E.M.; Holbert, R.C.; Pepine, C.J.; Raizada, M.K. Increased human intestinal barrier permeability plasma biomarkers zonulin and FABP2 correlated with plasma LPS and altered gut microbiome in anxiety or depression. Gut 2018, 67, 1555–1557. [Google Scholar] [CrossRef]
- Sarmin, M.; Begum, M.; Islam, F.; Afroze, F.; Shahrin, L.; Sharifuzzaman; Alam, T.; Shahid, A.; Ahmed, T.; Chisti, M.J. Factors associated with severe sepsis in diarrheal adults and their outcome at an urban hospital, Bangladesh: A retrospective analysis. PLoS ONE 2021, 16, e0257596. [Google Scholar] [CrossRef]
- Maier, L.; Goemans, C.V.; Wirbel, J.; Kuhn, M.; Eberl, C.; Pruteanu, M.; Müller, P.; Garcia-Santamarina, S.; Cacace, E.; Zhang, B.; et al. Unravelling the collateral damage of antibiotics on gut bacteria. Nature 2021, 599, 120–124. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Liu, Y.; Menkiti, O.R.; Mei, J.; Dai, N.; O’Leary, C.E.; Oliver, P.M.; Kolls, J.K.; Weiser, J.N.; Worthen, G.S. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 2014, 20, 524–530. [Google Scholar] [CrossRef] [Green Version]
- Baggs, J.; Jernigan, J.A.; Halpin, A.L.; Epstein, L.; Hatfield, K.M.; McDonald, L.C. Risk of Subsequent Sepsis Within 90 Days After a Hospital Stay by Type of Antibiotic Exposure. Clin. Infect. Dis. 2018, 66, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Lawn, J.E.; Bianchi-Jassir, F.; Russell, N.J.; Kohli-Lynch, M.; Tann, C.J.; Hall, J.; Madrid, L.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Group B Streptococcal Disease Worldwide for Pregnant Women, Stillbirths, and Children: Why, What, and How to Undertake Estimates? Clin. Infect. Dis. 2017, 65 (Suppl. 2), S89–S99. [Google Scholar] [CrossRef] [Green Version]
- Stoll, B.J.; Hansen, N.I.; Sánchez, P.J.; Faix, R.G.; Poindexter, B.B.; Van Meurs, K.P.; Bizzarro, M.J.; Goldberg, R.N.; Frantz, I.D., 3rd; Hale, E.C.; et al. Early onset neonatal sepsis: The burden of group B Streptococcal and E. coli disease continues. Pediatrics 2011, 127, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Sorbara, M.T.; Dubin, K.; Littmann, E.R.; Moody, T.U.; Fontana, E.; Seok, R.; Leiner, I.M.; Taur, Y.; Peled, J.U.; van den Brink, M.R.M.; et al. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J. Exp. Med. 2019, 216, 84–98. [Google Scholar] [CrossRef]
- Tanaka, S.; Couret, D.; Tran-Dinh, A.; Duranteau, J.; Montravers, P.; Schwendeman, A.; Meilhac, O. High-density lipoproteins during sepsis: From bench to bedside. Crit. Care 2020, 24, 134. [Google Scholar] [CrossRef] [Green Version]
- Caraballo, C.; Jaimes, F. Organ Dysfunction in Sepsis: An Ominous Trajectory From Infection To Death. Yale J. Biol. Med. 2019, 92, 629–640. [Google Scholar]
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66. [Google Scholar] [CrossRef]
- Duan, Y.; Chu, H.; Brandl, K.; Jiang, L.; Zeng, S.; Meshgin, N.; Papachristoforou, E.; Argemi, J.; Mendes, B.G.; Wang, Y.; et al. CRIg on liver macrophages clears pathobionts and protects against alcoholic liver disease. Nat. Commun. 2021, 12, 7172. [Google Scholar] [CrossRef]
- Bateman, R.M.; Sharpe, M.D.; Jagger, J.E.; Ellis, C.G.; Solé-Violán, J.; López-Rodríguez, M.; Herrera-Ramos, E.; Ruíz-Hernández, J. 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15–18 March 2016. Crit. Care 2016, 20 (Suppl. 2), 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruemmele, P.; Hofstaedter, F.; Gelbmann, C.M. Secondary sclerosing cholangitis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonça, A.; Bruining, H.; Reinhart, C.K.; Suter, P.M.; Thijs, L.G. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996, 22, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Dizier, S.; Forel, J.M.; Ayzac, L.; Richard, J.C.; Hraiech, S.; Lehingue, S.; Loundou, A.; Roch, A.; Guerin, C.; Papazian, L. Early Hepatic Dysfunction Is Associated with a Worse Outcome in Patients Presenting with Acute Respiratory Distress Syndrome: A Post-Hoc Analysis of the ACURASYS and PROSEVA Studies. PLoS ONE 2015, 10, e0144278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recknagel, P.; Gonnert, F.A.; Westermann, M.; Lambeck, S.; Lupp, A.; Rudiger, A.; Dyson, A.; Carré, J.E.; Kortgen, A.; Krafft, C.; et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: Experimental studies in rodent models of peritonitis. PLoS Med. 2012, 9, e1001338. [Google Scholar] [CrossRef]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S.; et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.; Schmidt, T.S.B.; Bengs, S.; Poveda, L.; Opitz, L.; Atrott, K.; Stanzel, C.; Biedermann, L.; Rehman, A.; Jonas, D.; et al. Effects of oral antibiotics and isotretinoin on the murine gut microbiota. Int. J. Antimicrob. Agents 2017, 50, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Wada, T.; Mizugaki, A.; Oda, Y.; Kayano, K.; Yamakawa, K.; Tanaka, S. Protocol for a Sepsis Model Utilizing Fecal Suspension in Mice: Fecal Suspension Intraperitoneal Injection Model. Front. Med. 2022, 9, 765805. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017. [Google Scholar] [CrossRef]
- Ellermann, M.; Pacheco, A.R.; Jimenez, A.G.; Russell, R.M.; Cuesta, S.; Kumar, A.; Zhu, W.; Vale, G.; Martin, S.A.; Raj, P.; et al. Endocannabinoids Inhibit the Induction of Virulence in Enteric Pathogens. Cell 2020, 183, 650–665.e15. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020, 28, 519–522. [Google Scholar] [CrossRef]
- Sun, D.; Bai, R.; Zhou, W.; Yao, Z.; Liu, Y.; Tang, S.; Ge, X.; Luo, L.; Luo, C.; Hu, G.F.; et al. Angiogenin maintains gut microbe homeostasis by balancing α-Proteobacteria and Lachnospiraceae. Gut 2021, 70, 666–676. [Google Scholar] [CrossRef]
- He, Z.; Ma, Y.; Yang, S.; Zhang, S.; Liu, S.; Xiao, J.; Wang, Y.; Wang, W.; Yang, H.; Li, S.; et al. Gut microbiota-derived ursodeoxycholic acid from neonatal dairy calves improves intestinal homeostasis and colitis to attenuate extended-spectrum β-lactamase-producing enteroaggregative Escherichia coli infection. Microbiome 2022, 10, 79. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agudelo-Ochoa, G.M.; Valdés-Duque, B.E.; Giraldo-Giraldo, N.A.; Jaillier-Ramírez, A.M.; Giraldo-Villa, A.; Acevedo-Castaño, I.; Yepes-Molina, M.A.; Barbosa-Barbosa, J.; Benítez-Paéz, A. Gut microbiota profiles in critically ill patients, potential biomarkers and risk variables for sepsis. Gut Microbes 2020, 12, 1707610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, B.W.; Wiersinga, W.J. The role of the gut microbiota in sepsis. Lancet Gastroenterol. Hepatol. 2017, 2, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Vich Vila, A.; Collij, V.; Sanna, S.; Sinha, T.; Imhann, F.; Bourgonje, A.R.; Mujagic, Z.; Jonkers, D.; Masclee, A.A.M.; Fu, J.; et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 2020, 11, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidi, A.; Ebadi, M.; Rehman, T.U.; Elhusseini, H.; Nalluri, H.; Kaiser, T.; Holtan, S.G.; Khoruts, A.; Weisdorf, D.J.; Staley, C. Gut microbiota response to antibiotics is personalized and depends on baseline microbiota. Microbiome 2021, 9, 211. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Wei, S.; Mortensen, M.S.; Stokholm, J.; Brejnrod, A.D.; Thorsen, J.; Rasmussen, M.A.; Trivedi, U.; Bisgaard, H.; Sørensen, S.J. Short- and long-term impacts of azithromycin treatment on the gut microbiota in children: A double-blind, randomized, placebo-controlled trial. EBioMedicine 2018, 38, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.M.; Chen, C.C.; Chang, C.M.; Fang, Y.J.; Bair, M.J.; Chen, P.Y.; Chang, C.Y.; Hsu, Y.C.; Chen, M.J.; Chen, C.C.; et al. Long-term changes of gut microbiota, antibiotic resistance, and metabolic parameters after Helicobacter pylori eradication: A multicentre, open-label, randomised trial. Lancet Infect. Dis. 2019, 19, 1109–1120. [Google Scholar] [CrossRef]
- Mirsepasi-Lauridsen, H.C.; Vallance, B.A.; Krogfelt, K.A.; Petersen, A.M. Escherichia coli Pathobionts Associated with Inflammatory Bowel Disease. Clin. Microbiol. Rev. 2019, 32, e00060-18. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Beyer, D.; Hoff, J.; Sommerfeld, O.; Zipprich, A.; Gaßler, N.; Press, A.T. The liver in sepsis: Molecular mechanism of liver failure and their potential for clinical translation. Mol. Med. 2022, 28, 84. [Google Scholar] [CrossRef]
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010, 207, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Zeller, J.; Bogner, B.; Kiefer, J.; Braig, D.; Winninger, O.; Fricke, M.; Karasu, E.; Peter, K.; Huber-Lang, M.; Eisenhardt, S.U. CRP Enhances the Innate Killing Mechanisms Phagocytosis and ROS Formation in a Conformation and Complement-Dependent Manner. Front. Immunol. 2021, 12, 721887. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020, 180, 79–91.e16. [Google Scholar] [CrossRef]
- Weersink, R.A.; Alvarez-Alvarez, I.; Medina-Cáliz, I.; Sanabria-Cabrera, J.; Robles-Díaz, M.; Ortega-Alonso, A.; García-Cortés, M.; Bonilla, E.; Niu, H.; Soriano, G.; et al. Clinical Characteristics and Outcome of Drug-Induced Liver Injury in the Older Patients: From the Young-Old to the Oldest-Old. Clin. Pharmacol. Ther. 2021, 109, 1147–1158. [Google Scholar] [CrossRef]
- Graversen, K.B.; Bahl, M.I.; Larsen, J.M.; Ballegaard, A.R.; Licht, T.R.; Bøgh, K.L. Short-Term Amoxicillin-Induced Perturbation of the Gut Microbiota Promotes Acute Intestinal Immune Regulation in Brown Norway Rats. Front. Microbiol. 2020, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Yu, Y.; Sun, R.; Huang, J.; Liu, L.; Yang, Y.; Rui, T.; Sun, B. Identification and characterization of neutrophil heterogeneity in sepsis. Crit. Care 2021, 25, 50. [Google Scholar] [CrossRef]
- Stanley, D.; Mason, L.J.; Mackin, K.E.; Srikhanta, Y.N.; Lyras, D.; Prakash, M.D.; Nurgali, K.; Venegas, A.; Hill, M.D.; Moore, R.J.; et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 2016, 22, 1277–1284. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and prebiotics in intestinal health and disease: From biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; de Vos, A.F.; de Beer, R.; Wieland, C.W.; Roelofs, J.J.; Woods, D.E.; van der Poll, T. Inflammation patterns induced by different Burkholderia species in mice. Cell Microbiol. 2008, 10, 81–87. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Ma, J.; Liu, H.; Luo, Y.; Deng, H.; Wang, D.; Wang, F.; Zhang, P. The Gut Microbiota Contributes to Systemic Responses and Liver Injury in Gut-Derived Sepsis. Microorganisms 2023, 11, 1741. https://doi.org/10.3390/microorganisms11071741
Zhao M, Ma J, Liu H, Luo Y, Deng H, Wang D, Wang F, Zhang P. The Gut Microbiota Contributes to Systemic Responses and Liver Injury in Gut-Derived Sepsis. Microorganisms. 2023; 11(7):1741. https://doi.org/10.3390/microorganisms11071741
Chicago/Turabian StyleZhao, Meiqi, Jiajia Ma, Huiru Liu, Ying Luo, Huiting Deng, Dandan Wang, Fengmei Wang, and Peng Zhang. 2023. "The Gut Microbiota Contributes to Systemic Responses and Liver Injury in Gut-Derived Sepsis" Microorganisms 11, no. 7: 1741. https://doi.org/10.3390/microorganisms11071741