
Rapid Identification of Lineage and Drug Resistance in Clinical Samples of Mycobacterium tuberculosis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
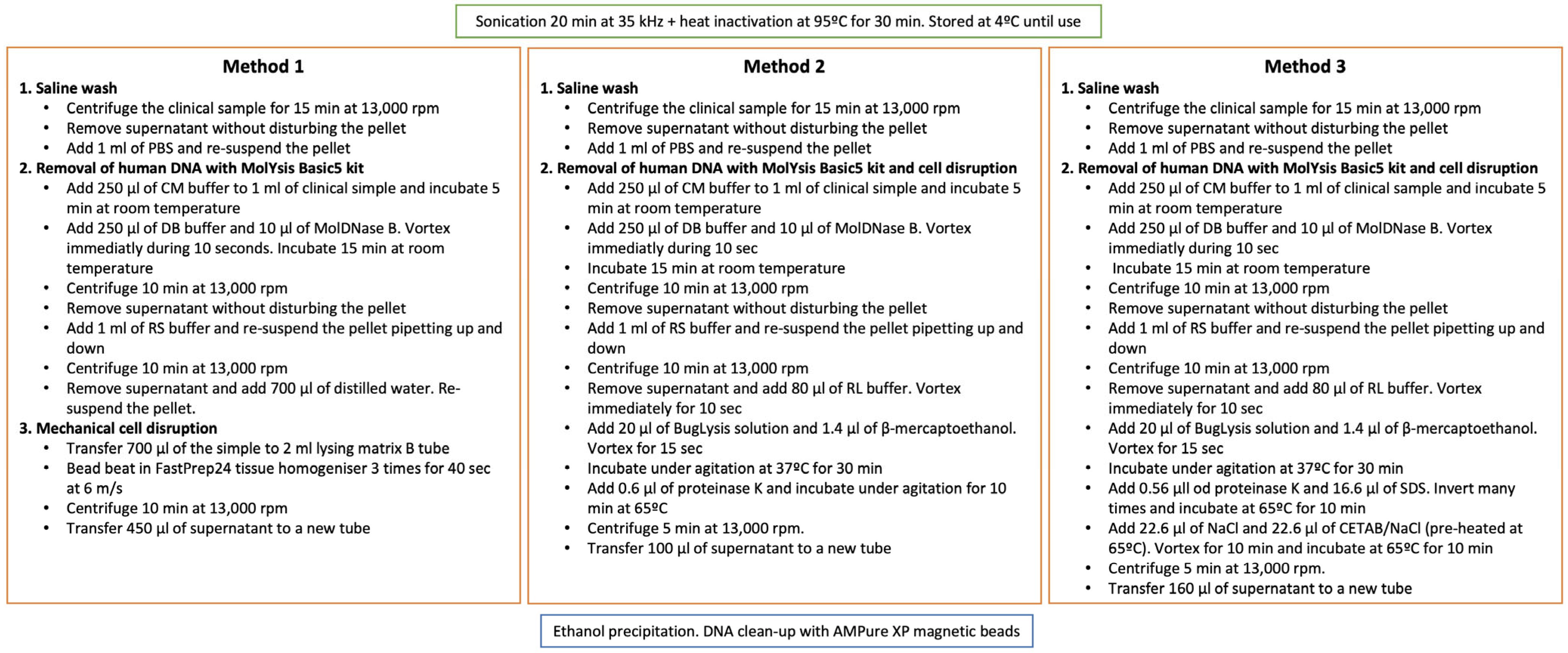
2.1. Set-Up of a DNA Extraction Method from Clinical Samples
2.2. Samples Analysed by AmpliSeq Technology
2.3. AmpliSeq Technology
2.4. Bioinformatic Analysis
3. Results
3.1. Set-Up of a DNA Extraction Method from Clinical Samples
3.2. AmpliSeq Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Votintseva, A.A.; Pankhurst, L.J.; Anson, L.W.; Morgan, M.R.; Gascoyne-Binzi, D.; Walker, T.M.; Quan, T.P.; Wyllie, D.H.; Del Ojo Elias, C.; Wilcox, M.; et al. Mycobacterial DNA extraction for whole-genome sequencing from early positive liquid (MGIT) cultures. J. Clin. Microbiol. 2015, 53, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Votintseva, A.A.; Bradley, P.; Pankhurst, L.; Del Ojo Elias, C.; Loose, M.; Nilgiriwala, K.; Chatterjee, A.; Smith, E.G.; Sanderson, N.; Walker, T.M.; et al. Same-Day Diagnostic and Surveillance Data for Tuberculosis via Whole-Genome Sequencing of Direct Respiratory Samples. J. Clin. Microbiol. 2017, 55, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Bryant, J.M.; Einer-Jensen, K.; Holdstock, J.; Houniet, D.T.; Chan, J.Z.M.; Depledge, D.P.; Nikolayevskyy, V.; Broda, A.; Stone, M.J.; et al. Rapid Whole-Genome Sequencing of Mycobacterium tuberculosis Isolates Directly from Clinical Samples. J. Clin. Microbiol. 2015, 53, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.M.; Burgess, C.; Williams, R.; Gorton, R.; Booth, H.; Brown, J.; Bryant, J.M.; Chan, J.; Creer, D.; Holdstock, J.; et al. Direct Whole-Genome Sequencing of Sputum Accurately Identifies Drug-Resistant Mycobacterium tuberculosis Faster than MGIT Culture Sequencing. J. Clin. Microbiol. 2018, 56, e00666-18. [Google Scholar] [CrossRef]
- Bonnet, I.; Enouf, V.; Morel, F.; Ok, V.; Jaffré, J.; Jarlier, V.; Aubry, A.; Robert, J.; Sougakoff, W. A Comprehensive Evaluation of GeneLEAD VIII DNA Platform Combined to Deeplex Myc-TB® Assay to Detect in 8 Days Drug Resistance to 13 Antituberculous Drugs and Transmission of Mycobacterium tuberculosis Complex Directly From Clinical Samples. Front. Cell. Infect. Microbiol. 2021, 11, 707244. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, D.M.; Cabibbe, A.M.; De Filippo, M.R.; Trovato, A.; Simonetti, T.; Rossolini, G.M.; Tortoli, E. Use of WGS in Mycobacterium tuberculosis routine diagnosis. Int. J. Mycobacteriol. 2016, 5, S252–S253. [Google Scholar] [CrossRef]
- van Soolingen, D.; de Haas, P.E.; Hermans, P.W.; van Embden, J.D. DNA fingerprinting of Mycobacterium tuberculosis. Methods Enzymol. 1994, 235, 196–205. [Google Scholar] [PubMed]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Lawn, S.D.; Nicol, M.P. Xpert® MTB/RIF assay: Development, evaluation and implementation of a new rapid molecular diagnostic for tuberculosis and rifampicin resistance. Future Microbiol. 2011, 6, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Theron, G.; Peter, J.; Richardson, M.; Barnard, M.; Donegan, S.; Warren, R.; Steingart, K.R.; Dheda, K. The diagnostic accuracy of the GenoType® MTBDRsl assay for the detection of resistance to second-line anti-tuberculosis drugs. Cochrane Database Syst. Rev. 2014, 10, CD010705. [Google Scholar] [CrossRef]
- Saha, A.; Vaidya, P.J.; Chavhan, V.B.; Pandey, K.V.; Kate, A.H.; Chhajed, P.N. Inconsistency in the reporting of antitubercular drug susceptibility tests in an endemic region. Lung India Off. Organ Indian Chest Soc. 2017, 34, 427–429. [Google Scholar] [CrossRef]
- Cabibbe, A.M.; Spitaleri, A.; Battaglia, S.; Colman, R.E.; Suresh, A.; Uplekar, S.; Rodwell, T.C.; Cirillo, D.M. Application of Targeted Next-Generation Sequencing Assay on a Portable Sequencing Platform for Culture-Free Detection of Drug-Resistant Tuberculosis from Clinical Samples. J. Clin. Microbiol. 2020, 58, e00632-20. [Google Scholar] [CrossRef] [PubMed]
- Simner, P.J.; Salzberg, S.L. The Human “Contaminome” and Understanding Infectious Disease. N. Engl. J. Med. 2022, 387, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Bakuła, Z.; Napiórkowska, A.; Bielecki, J.; Augustynowicz-Kopeć, E.; Zwolska, Z.; Jagielski, T. Mutations in the embB gene and their association with ethambutol resistance in multidrug-resistant Mycobacterium tuberculosis clinical isolates from Poland. Biomed Res. Int. 2013, 2013, 167954. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Levin, B.R. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 1999, 2, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2011, 44, 106–110. [Google Scholar] [CrossRef] [PubMed]
- García De Viedma, D.; Pérez-Lago, L. The Evolution of Genotyping Strategies To Detect, Analyze, and Control Transmission of Tuberculosis. Microbiol. Spectr. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Ates, L.S.; Dippenaar, A.; Ummels, R.; Piersma, S.R.; Van Der Woude, A.D.; Van Der Kuij, K.; Le Chevalier, F.; Mata-Espinosa, D.; Barrios-Payán, J.; Marquina-Castillo, B.; et al. Mutations in ppe38 block PE-PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat. Microbiol. 2018, 3, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Borrell, S.; Gagneux, S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1456–1466. [Google Scholar]


{kind=link}
| Sample | ng/μL | Kind of Sample | BK | PCR | Sample | ng/μL | Kind of Sample |
|---|---|---|---|---|---|---|---|
| 96 | LOW | Sputum | + | Yes | MTB-1 | 12.5 | Bacterial culture |
| 91 | LOW | Sputum | + | Yes | MTB-2 | 37.6 | Bacterial culture |
| 955 | 8.14 | Sputum | + | Yes | MTB-3 | 23.9 | Bacterial culture |
| 52 | 2.28 | Sputum | + | Yes | MTB-4 | 40.6 | Bacterial culture |
| 344 | 0.488 | Sputum | + | Yes | MTB-5 | 55.4 | Bacterial culture |
| 692 | 0.598 | Sputum | + | Yes | MTB-6 | 43 | Bacterial culture |
| 785 | 3.12 | Sputum | + | Yes | MTB-7 | 11.2 | Bacterial culture |
| 532 | 0.162 | Sputum | − | Yes | MTB-8 | 326 | Bacterial culture |
| 952 | 6 | Sputum | + | Yes | MTB-9 | 35.8 | Bacterial culture |
| 684 | 8.5 | Sputum | + | Yes | MTB-10 | 8.04 | Bacterial culture |
| 879 | LOW | Pleural fluid | + | Yes | MTB-11 | 9.98 | Bacterial culture |
| 635 | 2.52 | Biopsy | + | Yes | MTB-12 | 7.74 | Bacterial culture |
| 942 | LOW | Biopsy | − | No | MTB-13 | 6.56 | Bacterial culture |
| 217 | LOW | Sputum | + | Yes | MTB-14 | 2.66 | Bacterial culture |
| 659 | 5 | Sputum | + | Yes | MTB-15 | 2 | Bacterial culture |
| 388 | 2.18 | Sputum | + | Yes | MTB-16 | 1.56 | Bacterial culture |
| 658 | LOW | Sputum | + | Yes | MTB-17 | 20.6 | Bacterial culture |
| 275 | LOW | Sputum | + | Yes | MTB-18 | 11.8 | Bacterial culture |
| 698 | 5.72 | Sputum | + | Yes | MTB-19 | 51.6 | Bacterial culture |
| 052 | LOW | Sputum | + | Yes | MTB-20 | 2.86 | Bacterial culture |
| 542 | 1.67 | Sputum | + | Yes | MTB-21 | 17.6 | Bacterial culture |
| 315 | 0.68 | Sputum | + | Yes | MTB-22 | 10.3 | Bacterial culture |
| 40 | LOW | Sputum | + | Yes | MTB-23 | 20.7 | Bacterial culture |
| 988 | 0.254 | Sputum | − | Yes | MTB-24 | 8.4 | Bacterial culture |
| 212 | 0.368 | Sputum | + | Yes | MTB-25 | 2.46 | Bacterial culture |
| 140 | LOW | Sputum | + | Yes | MTB-26 | 40 | Bacterial culture |
| 381 | LOW | Sputum | + | Yes | MTB-27 | 23 | Bacterial culture |
| 786 | LOW | Sputum | + | Yes | MTB-28 | 2.96 | Bacterial culture |
| 644 | LOW | Sputum | + | Yes | MTB-29 | 6.2 | Bacterial culture |
| 912 | 0.482 | Sputum | + | Yes | MTB-30 | 43 | Bacterial culture |
| 120 | LOW | Sputum | + | Yes | MTB-31 | 4.82 | Bacterial culture |
| 270 | LOW | Biopsy | - | Yes | MTB-32 | 15.1 | Bacterial culture |
| 178 | 0.422 | Sputum | + | Yes | MTB-33 | 472 | Bacterial culture |
| 537 | 0.454 | Sputum | − | Yes | MTB-34 | 14.8 | Bacterial culture |
| 916 | 0.138 | Sputum | + | Yes | MTB-35 | 139 | Bacterial culture |
| 521 | 0.122 | Sputum | - | Yes | MTB-36 | 32.2 | Bacterial culture |
| 736 | 1.92 | Sputum | + | Yes | MTB-37 | 60.6 | Bacterial culture |
| 69 | LOW | Sputum | + | Yes | MTB-38 | 111 | Bacterial culture |
| 263 | 0.19 | Sputum | + | Yes | MTB-39 | 21.4 | Bacterial culture |
| 453 | 7.22 | Sputum | + | Yes | MTB-40 | 104 | Bacterial culture |
| 163 | 0.106 | Aspirate | + | No | MTB-41 | 70.2 | Bacterial culture |
| 667 | 0.162 | Aspirate | + | Yes | MTB-42 | 48.2 | Bacterial culture |
| 882 | 0.328 | Sputum | + | Yes | MTB-43 | 3.92 | Bacterial culture |
| 007 | LOW | Sputum | + | No | MTB-44 | 8.08 | Bacterial culture |
| 640 | LOW | Lavage | − | No | MTB-45 | 108.7 | Bacterial culture |
| 716 | LOW | Aspirate | − | No | MTB-46 | 6.12 | Bacterial culture |
| 907 | LOW | Pleural fluid | − | No | MTB-47 | 8.42 | Bacterial culture |
| 100 | 0.964 | Sputum | − | No | MTB-48 | 6.24 | Bacterial culture |
| 561 | LOW | Adeno puncture | − | No | MTB-49 | 2 | Bacterial culture |
| 327 | LOW | Aspirate | − | No | MTB-50 | 14.3 | Bacterial culture |
| 343 | LOW | Aspirate | − | No | MTB-51 | 0.744 | Bacterial culture |
| 590 | 0.114 | Sputum | − | No | MTB-52 | 1.292 | Bacterial culture |
| 169 | LOW | Pleural fluid | − | No | |||
| 273 | 1.54 | Sputum | + | Yes | |||
| 884 | 0.152 | Lavage | − | No | |||
| 295 | 0.202 | Sputum | − | No | |||
| 318 | LOW | Aspirate | + | No | |||
| 473 | 0.84 | Sputum | − | No | |||
| 366 | LOW | Pleural fluid | − | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comín, J.; Viñuelas, J.; Lafoz, C.; Cebollada, A.; Ibarz, D.; Iglesias, M.-J.; Samper, S. Rapid Identification of Lineage and Drug Resistance in Clinical Samples of Mycobacterium tuberculosis. Microorganisms 2023, 11, 1467. https://doi.org/10.3390/microorganisms11061467
Comín J, Viñuelas J, Lafoz C, Cebollada A, Ibarz D, Iglesias M-J, Samper S. Rapid Identification of Lineage and Drug Resistance in Clinical Samples of Mycobacterium tuberculosis. Microorganisms. 2023; 11(6):1467. https://doi.org/10.3390/microorganisms11061467
Chicago/Turabian StyleComín, Jéssica, Jesús Viñuelas, Carmen Lafoz, Alberto Cebollada, Daniel Ibarz, María-José Iglesias, and Sofía Samper. 2023. "Rapid Identification of Lineage and Drug Resistance in Clinical Samples of Mycobacterium tuberculosis" Microorganisms 11, no. 6: 1467. https://doi.org/10.3390/microorganisms11061467