
HLA-Haplotypes Influence Microbiota Structure in Northwestern Mexican Schoolchildren Predisposed for Celiac Disease or Type 1 Diabetes
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genotyping and Risk for CD and T1D Identification
2.2. Autoantibody Detection
2.3. Microbiota and Diet Analysis
2.4. Bioinformatics and Statistical Analysis
3. Results
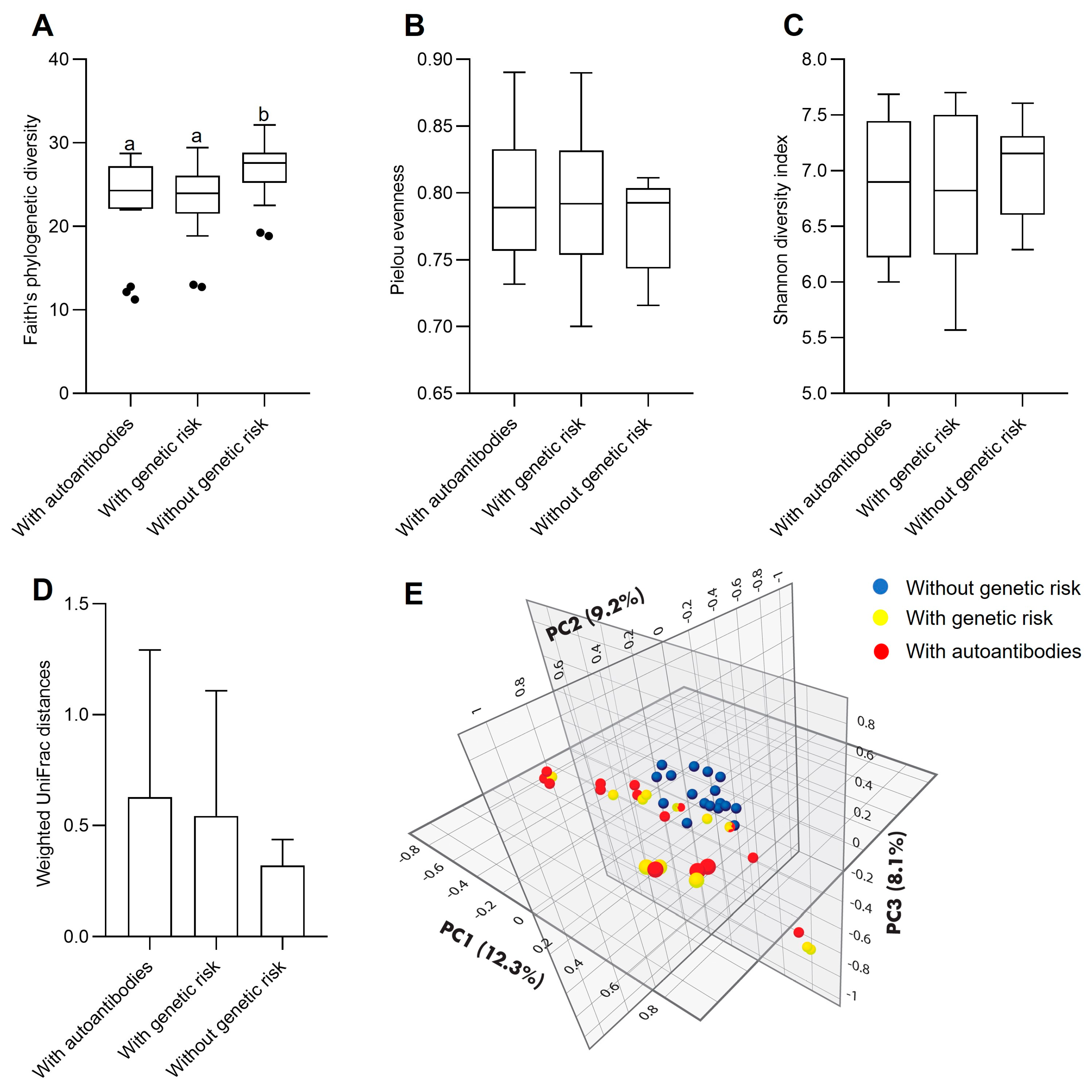
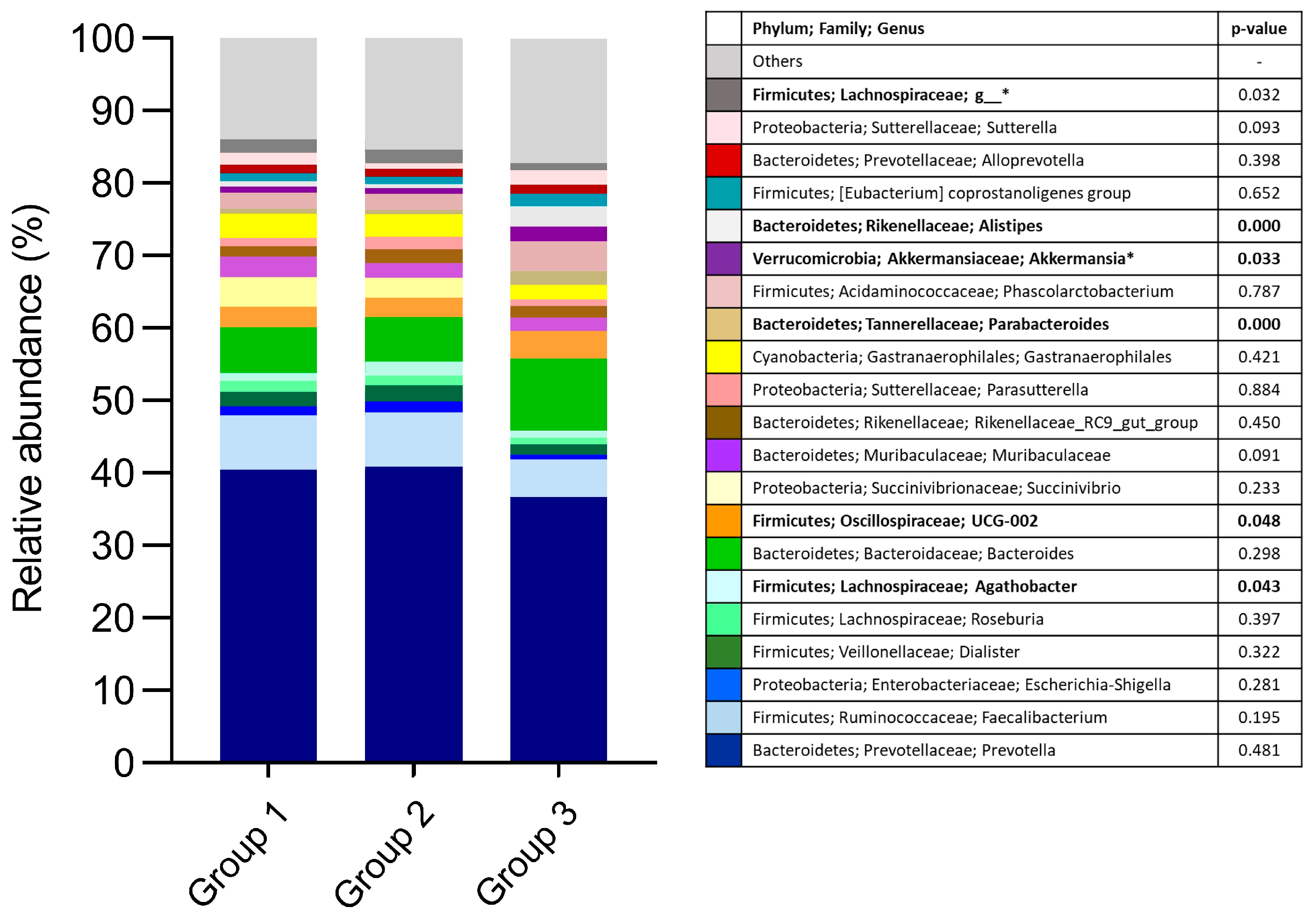
3.1. Fecal Bacterial Microbiota
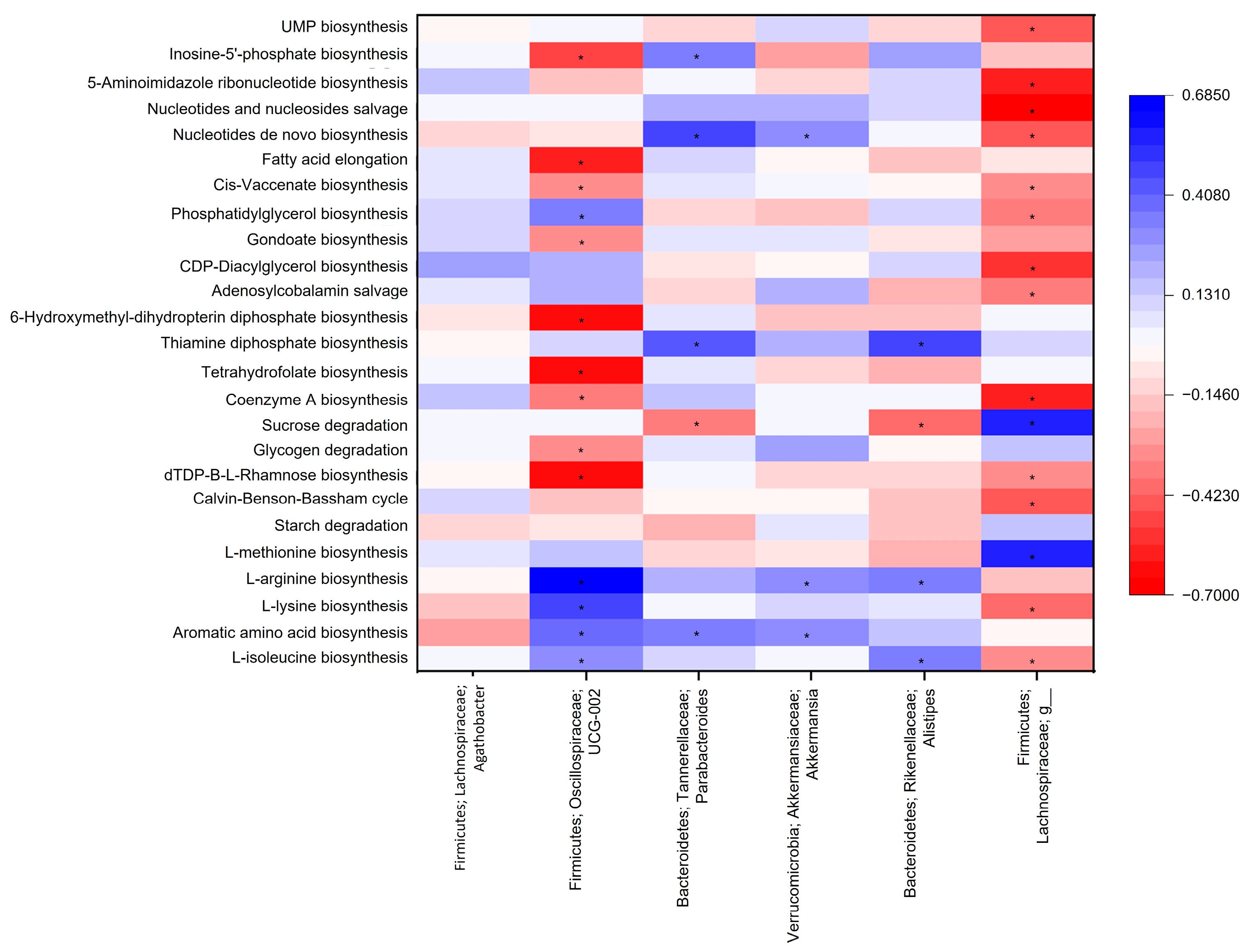
3.2. Microbiota Functional Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodwin, G. Type 1 Diabetes Mellitus and Celiac Disease: Distinct Autoimmune Disorders That Share Common Pathogenic Mechanisms. Horm. Res. Paediatr. 2019, 92, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Williams, E.E.; Lorca, G.L.; Norris, J.M.; Dunne, J.L. A Triple Threat? The Role of Diet, Nutrition, and the Microbiota in T1D Pathogenesis. Front. Nutr. 2021, 8, 600756. [Google Scholar] [CrossRef] [PubMed]
- Skoracka, K.; Hryhorowicz, S.; Rychter, A.M.; Ratajczak, A.E.; Szymczak-Tomczak, A.; Zawada, A.; Słomski, R.; Dobrowolska, A.; Krela-Kaźmierczak, I. Why are western diet and western lifestyle pro-inflammatory risk factors of celiac disease? Front. Nutr. 2023, 9, 1054089. [Google Scholar] [CrossRef] [PubMed]
- Calderón de la Barca, A.M.; Castillo-Fimbres, R.S.; Mejía-León, M.E.; Quihui-Cota, L.; Ochoa-Leyva, A.; Aguayo-Patrón, S.V. Enteric parasitic infection disturbs bacterial structure in Mexican children with autoantibodies for type 1 diabetes and/or celiac disease. Gut Pathog. 2020, 12, 37. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, R.; Han, B.; Sun, C.; Chen, R.; Wei, H.; Chen, L.; Du, H.; Li, G.; Yang, Y.; et al. Functional and metabolic alterations of gut microbiota in children with new-onset type 1 diabetes. Nat. Commun. 2022, 13, 6356. [Google Scholar] [CrossRef]
- Mejía-León, M.E.; Petrosino, J.F.; Ajami, N.J.; Domínguez-Bello, M.G.; Calderón de la Barca, A.M. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci. Rep. 2014, 4, 3814. [Google Scholar] [CrossRef]
- Leonard, M.M.; Valitutti, F.; Karathia, H.; Pujolassos, M.; Kenyon, V.; Fanelli, B.; Troisi, J.; Subramanian, P.; Camhi, S.; Colucci, A.; et al. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc. Natl. Acad. Sci. USA 2021, 118, e2020322118. [Google Scholar] [CrossRef]
- Biassoni, R.; Di Marco, E.; Squillario, M.; Barla, A.; Piccolo, G.; Ugolotti, E.; Gatti, C.; Minuto, N.; Patti, G.; Maghnie, M.; et al. Gut Microbiota in T1DM-Onset Pediatric Patients: Machine-Learning Algorithms to Classify Microorganisms as Disease Linked. J. Clin. Endocrinol. Metab. 2020, 105, dgaa407. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Instituto Nacional de Estadística y Geografía. Población Según Condición de Afiliación a Servicios de Salud y Sexo Por Entidad Federativa. 2020. Available online: https://inegi.org.mx/app/tabulados/interactivos/?pxq=Derechohabiencia_Derechohabiencia_01_3e83e8a1-690d-4cfb-8af0-a1e675979b3e (accessed on 7 April 2023).
- Ortega, M.I.; Sabo, S.; Aranda Gallegos, P.; De Zapien, J.E.; Zapien, A.; Portillo Abril, G.E.; Rosales, C. Agribusiness, Corporate Social Responsibility, and Health of Agricultural Migrant Workers. Front. Public Health 2016, 4, 54. [Google Scholar] [CrossRef]
- Aguayo-Patrón, S.; Beltrán-Sauceda, L.; Calderón de la Barca, A.M. A population-wide applicable HLA-DQ2 and DQ8 genotyping using DNA from dried blood spots and duplex allele-specific qPCR amplification. Scand. J. Clin. Lab. Investig. 2016, 76, 581–587. [Google Scholar] [CrossRef]
- Mejía-León, M.; Ruiz-Dyck, K.; de la Barca, A.C. HLA-DQ genetic risk gradient for type 1 diabetes and celiac disease in northwestern Mexico. Rev. Gastroenterol. México 2015, 80, 135–143. [Google Scholar] [CrossRef]
- Leonard, M.M.; Karathia, H.; Pujolassos, M.; Troisi, J.; Valitutti, F.; Subramanian, P.; Camhi, S.; Kenyon, V.; Colucci, A.; Serena, G.; et al. Multi-omics analysis reveals the influence of genetic and environmental risk factors on developing gut microbiota in infants at risk of celiac disease. Microbiome 2020, 8, 130. [Google Scholar] [CrossRef]
- Cabrera-Chávez, F.; Rouzaud-Sández, O.; Sotelo-Cruz, N.; Calderón de la Barca, A.M. Bovine milk caseins and transglutaminase-treated cereal prolamins are differentially recognized by IgA of celiac disease patients according to their age. J. Agric. Food Chem. 2009, 57, 3754–3759. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.I.; Quizán, P.T.; Morales, F.G.G.; Preciado, M. Cálculo de ingestión dietaria y coeficientes de adecuación a partir de registro de 24 horas y frecuencia de consumo de alimentos. In Estimación del Consumo de Alimentos; Cuaderno de trabajo No. 1; Serie de Evaluación del Consumo de Alimentos; Centro de Investigación en Alimentación y Desarrollo A.C.: Hermosillo, México, 1999. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut Microbiota Diversity and Human Diseases: Should We Reintroduce Key Predators in Our Ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Knight, R. Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev. 2008, 32, 557–578. [Google Scholar] [CrossRef]
- Olivares, M.; Neef, A.; Castillejo, G.; Palma, G.D.; Varea, V.; Capilla, A.; Palau, F.; Nova, E.; Marcos, A.; Polanco, I.; et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 2015, 64, 406–417. [Google Scholar] [CrossRef]
- Russell, J.T.; Roesch, L.F.W.; Ördberg, M.; Ilonen, J.; Atkinson, M.A.; Schatz, D.A.; Triplett, E.W.; Ludvigsson, J. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat. Commun. 2019, 10, 3621. [Google Scholar] [CrossRef] [PubMed]
- Marietta, E.; Rishi, A.; Taneja, V. Immunogenetic control of the intestinal microbiota. Immunology 2015, 145, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ni, J.J.; Han, B.X.; Yan, S.S.; Wei, X.T.; Feng, G.; Zhang, H.; Zhang, L.; Li, B.; Pei, Y.F. Causal Relationship Between Gut Microbiota and Autoimmune Diseases: A Two-Sample Mendelian Randomization Study. Front. Immunol. 2022, 12, 746998. [Google Scholar] [CrossRef] [PubMed]
- Krych, Ł.; Nielsen, D.S.; Hansen, A.K.; Hansen, C.H. Gut microbial markers are associated with diabetes onset, regulatory imbalance, and IFN-γ level in NOD mice. Gut Microbes 2015, 6, 101–109. [Google Scholar] [CrossRef]
- Del Chierico, F.; Conta, G.; Matteoli, M.C.; Fierabracci, A.; Reddel, S.; Macari, G.; Gardini, S.; Guarrasi, V.; Levi Mortera, S.; Marzano, V.; et al. Gut Microbiota Functional Traits, Blood pH, and Anti-GAD Antibodies Concur in the Clinical Characterization of T1D at Onset. Int. J. Mol. Sci. 2022, 23, 10256. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361. [Google Scholar] [CrossRef]
- Di Rienzi, S.C.; Britton, R.A. Adaptation of the Gut Microbiota to Modern Dietary Sugars and Sweeteners. Adv. Nutr. 2020, 11, 616–629. [Google Scholar] [CrossRef]
- Marrón-Ponce, J.A.; Sánchez-Pimienta, T.G.; Louzada, M.L.D.C.; Batis, C. Energy contribution of NOVA food groups and sociodemographic determinants of ultra-processed food consumption in the Mexican population. Public Health Nutr. 2018, 21, 87–93. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Bélteky, M.; Milletich, P.L.; Ahrens, A.P.; Triplett, E.W.; Ludvigsson., J. Infant gut microbiome composition correlated with type 1 diabetes acquisition in the general population: The ABIS study. Diabetologia 2023, in press. [Google Scholar] [CrossRef]
- El Mouzan, M.; Al-Hussaini, A.; Serena, G.; Assiri, A.; Al Sarkhy, A.; Al Mofarreh, M.; Alasmi, M.; Fasano, A. Microbiota profile of new-onset celiac disease in children in Saudi Arabia. Gut Pathog. 2022, 14, 37. [Google Scholar] [CrossRef]
- Di Biase, A.R.; Marasco, G.; Ravaioli, F.; Dajti, E.; Colecchia, L.; Righi, B.; D’Amico, V.; Festi, D.; Iughetti, L.; Colecchia, A. Gut microbiota signatures and clinical manifestations in celiac disease children at onset: A pilot study. J. Gastroenterol. Hepatol. 2020, 36, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Feng, S.; Arjan, N.; Chen, W. A next generation probiotic, Akkermansia muciniphila. Crit. Rev. Food Sci. Nutr. 2019, 59, 3227–3236. [Google Scholar] [CrossRef] [PubMed]
- Mønsted, M.Ø.; Falck, N.D.; Pedersen, K.; Buschard, K.; Holm, L.J.; Haupt-Jorgensen, M. Intestinal permeability in type 1 diabetes: An updated comprehensive overview. J. Autoimmun. 2021, 122, 102674. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves Miranda, M.C.; Pires Oliveira, R.; Torres, L.; Florini Aguiar, S.L.; Pinheiro-Rosa, N.; Lemos, L.; Guimarães, M.A.; Reis, D.; Silveira, T.; Ferreira, Ê.; et al. Frontline Science: Abnormalities in the gut mucosa of non-obese diabetic mice precede the onset of type 1 diabetes. J. Leukoc. Biol. 2019, 106, 513–529. [Google Scholar] [CrossRef]
- Raimondi, S.; Musmeci, E.; Candeliere, F.; Amaretti, A.; Rossi, M. Identification of mucin degraders of the human gut microbiota. Sci. Rep. 2021, 11, 11094. [Google Scholar] [CrossRef]
- Leth, M.L.; Pichler, M.J.; Abou Hachem, M. Butyrate-producing colonic clostridia: Picky glycan utilization specialists. Essays Biochem. 2022, 9, EBC20220125. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, L.; Wang, X.; Yi, Y.; Shan, Y.; Liu, B.; Zhou, Y.; Lü, X. Roles of intestinal Parabacteroides in human health and diseases. FEMS Microbiol. Lett. 2022, 369, fnac072. [Google Scholar] [CrossRef]
- Girdhar, K.; Huang, Q.; Chow, I.T.; Vatanen, T.; Brady, C.; Raisingani, A.; Autissier, P.; Atkinson, M.A.; Kwok, W.W.; Kahn, C.R.; et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2120028119. [Google Scholar] [CrossRef]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Group 1 | Group 2 | Group 3 | p-Value |
|---|---|---|---|---|
| (n = 18) | (n = 18) | (n = 17) | ||
| Sex, female, % | 72.2 | 66.7 | 64.7 | 0.884 |
| Age, mean (SE), years | 9.28 (0.39) | 9.56 (0.42) | 9.59 (0.30) | 0.813 |
| Weight, median (IQR), kg | 33.70 (26.14–41.15) | 32.45 (24.97–42.40) | 36.65 (31.03–48.15) | 0.299 |
| Height, median (IQR), cm | 1.37 (1.28–1.49) | 1.39 (1.31–1.47) | 1.37 (1.30–1.48) | 0.969 |
| BMI/age, mean (SE), Z-Score | 0.69 (1.43) | 0.64 (1.53) | 1.43 (1.13) | 0.173 |
| Diet | ||||
| Total energy intake, mean (SE), kcal | 1715.25 (61.84) | 1675.17 (119.71) | 1533.31 (76.35) | 0.327 |
| Carbohydrates intake, mean (SE), % * | 54.80 (2.16) | 56.82 (1.98) | 53.31 (2.31) | 0.517 |
| Protein intake, mean (SE), % * | 11.82 (0.84) | 11.45 (0.79) | 11.64 (0.67) | 0.058 |
| Fat intake, mean (SE), % * | 33.98 (1.97) | 33.63 (1.62) | 36.59 (1.86) | 0.466 |
| Saturated fat intake, mean (SE), % * | 10.46 (0.59) | 9.65 (0.63) | 11.84 (1.01) | 0.130 |
| Fiber intake, median (IQR), g/day | 16.96 (12.60–20.74) | 15.83 (10.24–24.34) | 11.72 (9.16–19.18) | 0.193 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguayo-Patrón, S.V.; Trujillo-Rivera, O.A.; Cornejo-Granados, F.; Ochoa-Leyva, A.; Calderón de la Barca, A.M. HLA-Haplotypes Influence Microbiota Structure in Northwestern Mexican Schoolchildren Predisposed for Celiac Disease or Type 1 Diabetes. Microorganisms 2023, 11, 1412. https://doi.org/10.3390/microorganisms11061412
Aguayo-Patrón SV, Trujillo-Rivera OA, Cornejo-Granados F, Ochoa-Leyva A, Calderón de la Barca AM. HLA-Haplotypes Influence Microbiota Structure in Northwestern Mexican Schoolchildren Predisposed for Celiac Disease or Type 1 Diabetes. Microorganisms. 2023; 11(6):1412. https://doi.org/10.3390/microorganisms11061412
Chicago/Turabian StyleAguayo-Patrón, Sandra V., Omar A. Trujillo-Rivera, Fernanda Cornejo-Granados, Adrian Ochoa-Leyva, and Ana M. Calderón de la Barca. 2023. "HLA-Haplotypes Influence Microbiota Structure in Northwestern Mexican Schoolchildren Predisposed for Celiac Disease or Type 1 Diabetes" Microorganisms 11, no. 6: 1412. https://doi.org/10.3390/microorganisms11061412