

Gut Microbiome in Post-COVID-19 Patients Is Linked to Immune and Cardiovascular Health Status but Not COVID-19 Severity
 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Trial Information
2.2. General Recruitment Criteria
- Confirmed COVID-19 diagnosis based on:
- ○
- A medical record of a positive RT-PCR test result for SARS-CoV-2; or
- ○
- IgM and IgG antibody titers against SARS-CoV-2 in the serum, indicating that the participant has been previously infected with SARS-CoV-2.
- No history of clinical symptoms associated with COVID-19 in the past six months, including fever (body temperature above 37.5 degrees Celsius/99.5 degrees Fahrenheit), shortness of breath, smell and taste dysfunctions, diarrhea, and coughing.
- Previous close contact for at least three days without personal protective equipment with individuals who exhibited COVID-19 symptoms (at home, in the workplace, etc.)
- No IgM and IgG antibody titers against SARS-CoV-2 in the serum, indicating that the participant has not been previously infected with SARS-CoV-2
- No history of clinical symptoms associated with COVID-19 for the past six months, including fever (body temperature above 37.5 degrees Celsius/99.5 degrees Fahrenheit), shortness of breath, smell and taste dysfunctions, diarrhea, and coughing.
- Confirmed COVID-19 diagnosis based on:
- ○
- A medical record of a positive RT-PCR test result for SARS-CoV-2; or
- ○
- IgM and IgG antibody titers against SARS-CoV-2 in the serum, indicating that the participant has been previously infected with SARS-CoV-2.
- Inpatient treatment of severe COVID-19
- Respiratory rate > 30 breaths/min
- Blood oxygen saturation (SpO2) ≤ 93%
- PaO2/FiO2 < 300
- Computed tomography (CT) findings of lung damage > 50%
- Septic shock
- Multiple organ failure
- Cytokine storm
- Pregnancy
- Refusal to participate
- Prior COVID-19 vaccination
- Signing of an informed consent form
- Express pregnancy test (for women of childbearing age)
- Measurement of IgM and IgG antibody titers against SARS-CoV-2
- Intake of medical and epidemiological histories
- An extensive survey and intake of detailed life and medical histories
- Physical examination, including measurement of arterial blood pressure (ABP), heart rate (HR), respiratory rate (RR), and anthropometric status indicators including hip and waist circumference and body mass index (BMI)
- Complete blood count with white blood cell differential
- Blood chemistry test, including homeostasis, immune status, level of sex hormones, and hormonal markers of metabolic dysfunctions
- Bioelectrical impedance analysis
- Stool sampling for gut microbiota analysis by 16S rRNA gene sequencing (Group A, 45 samples; Group N, 50 samples; Group S, 88 samples)
- Express smell test
- Chest CT
- Spirometry
- Speckle tracking echocardiography
2.3. Sample Collection and Processing
2.4. Data Analysis
- Laboratory tests related to immune status (n = 18)
- Clinical cardiovascular markers (n = 21)
- Laboratory markers of endothelial dysfunction (n = 5)
- Blood metabolites (n = 21)
3. Results
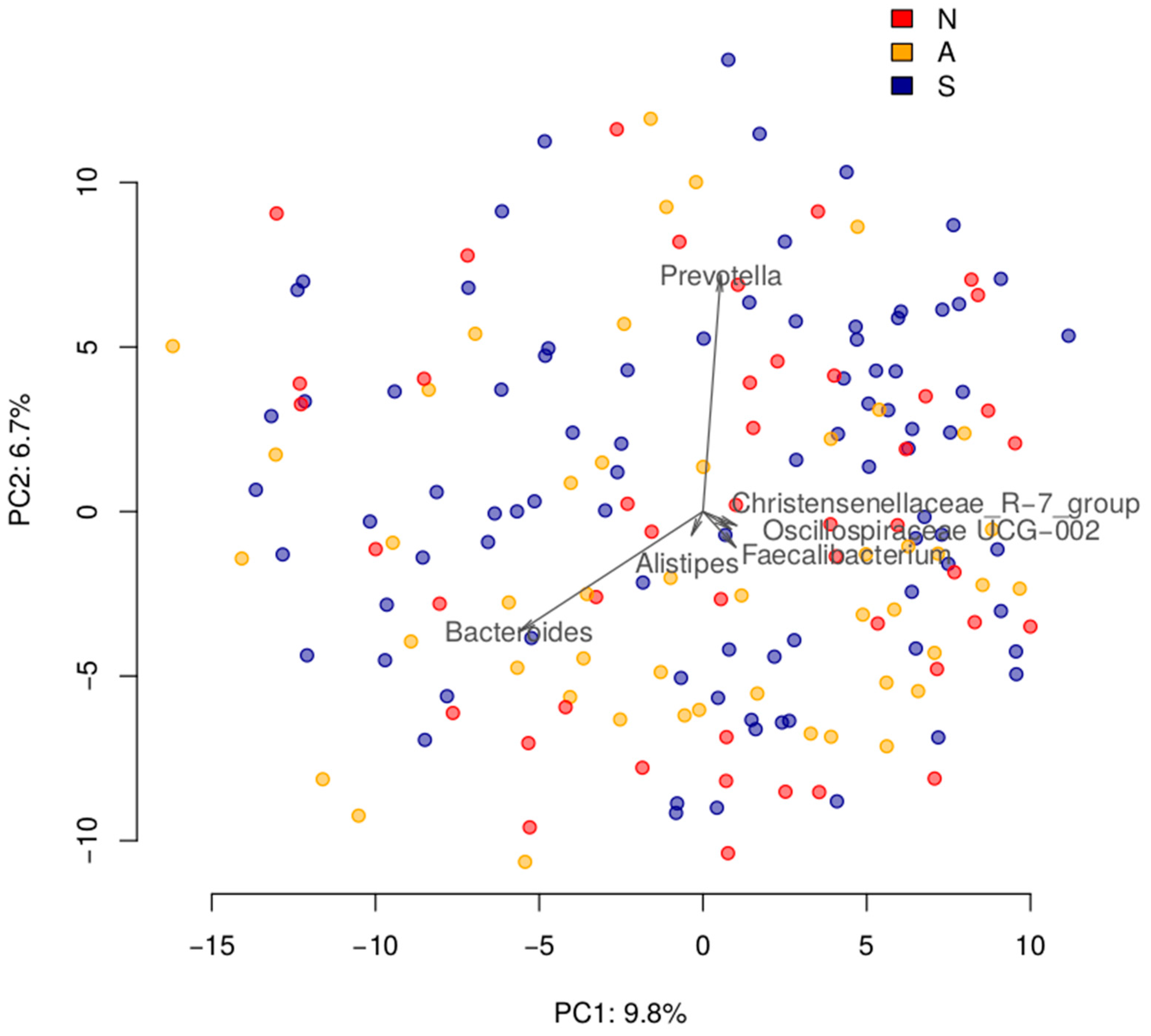
3.1. Gut Microbiome Composition Is Not Associated with the Severity of Disease after Three Months
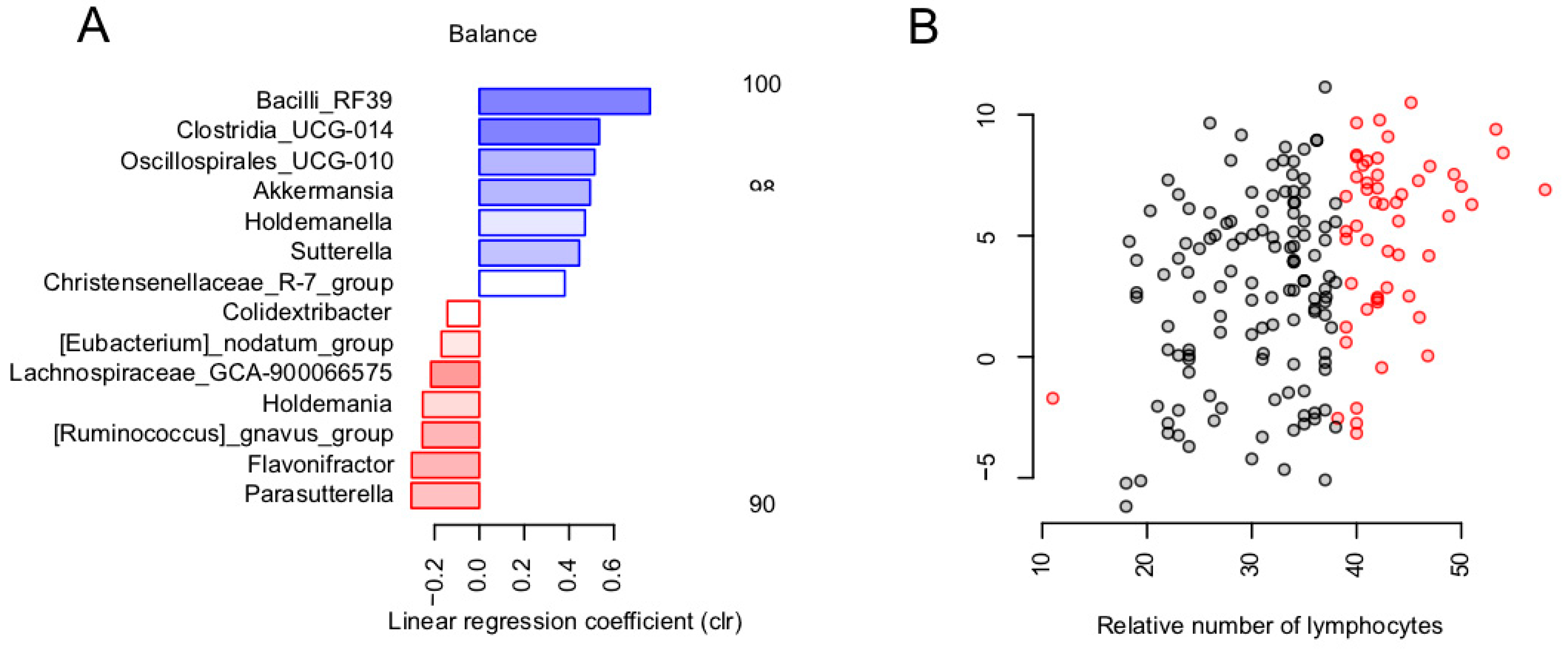
3.2. Balance of Specific Bacterial Taxa Is Linked to Immunity via Relative Level of Lymphocytes
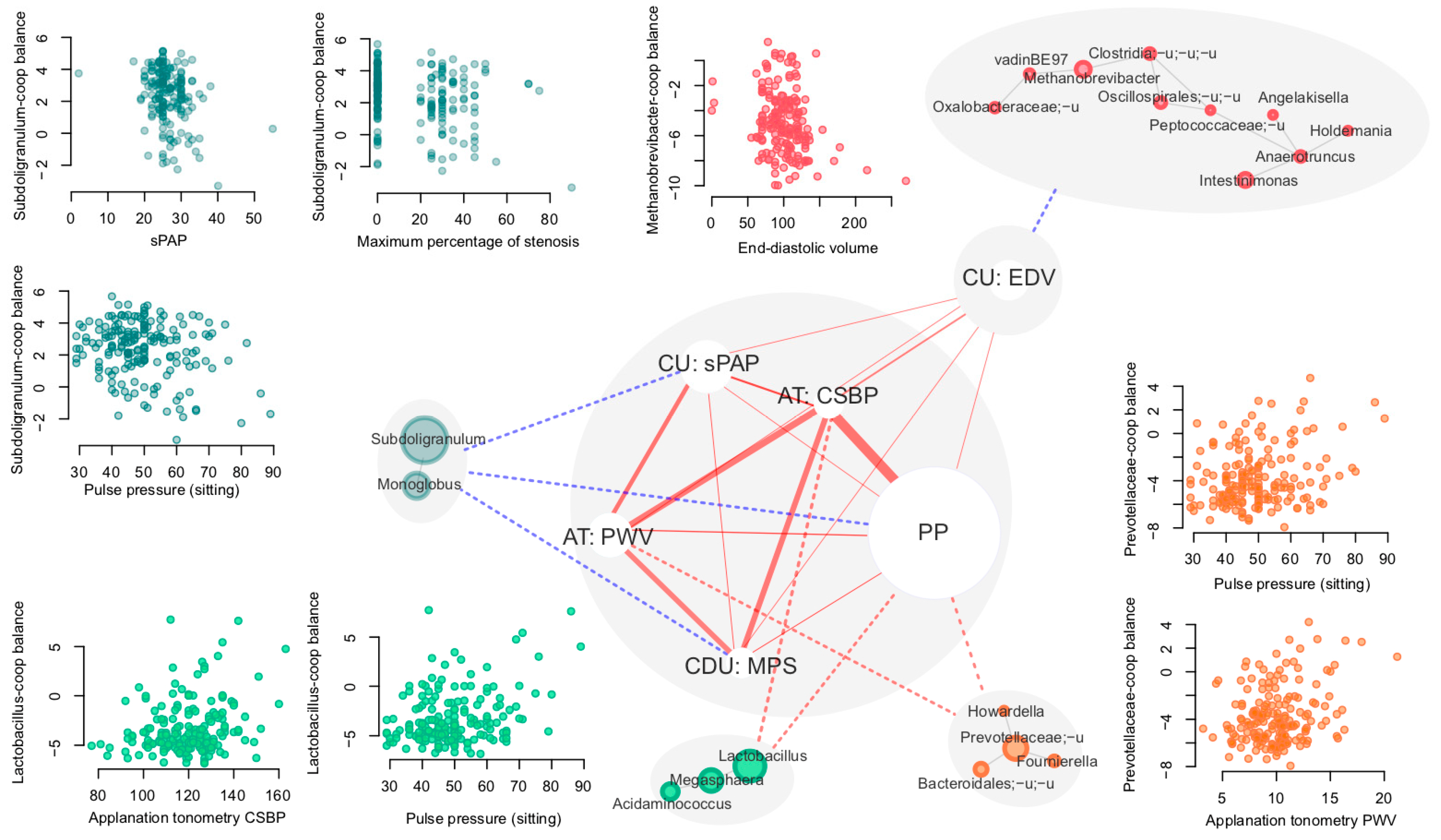
3.3. Links between Microbiome and Clinical Cardiovascular and Endothelial Dysfunction Markers
- -
- Vascular endothelial soluble growth factor receptor 1 (VEGF-R1)
- -
- Intercellular adhesion molecule 1 (ICAM-1)
- -
- Vascular endothelial adhesion molecule type 1 (VCAM-1)
- -
- E-selectin
- -
- Von Willebrand factor
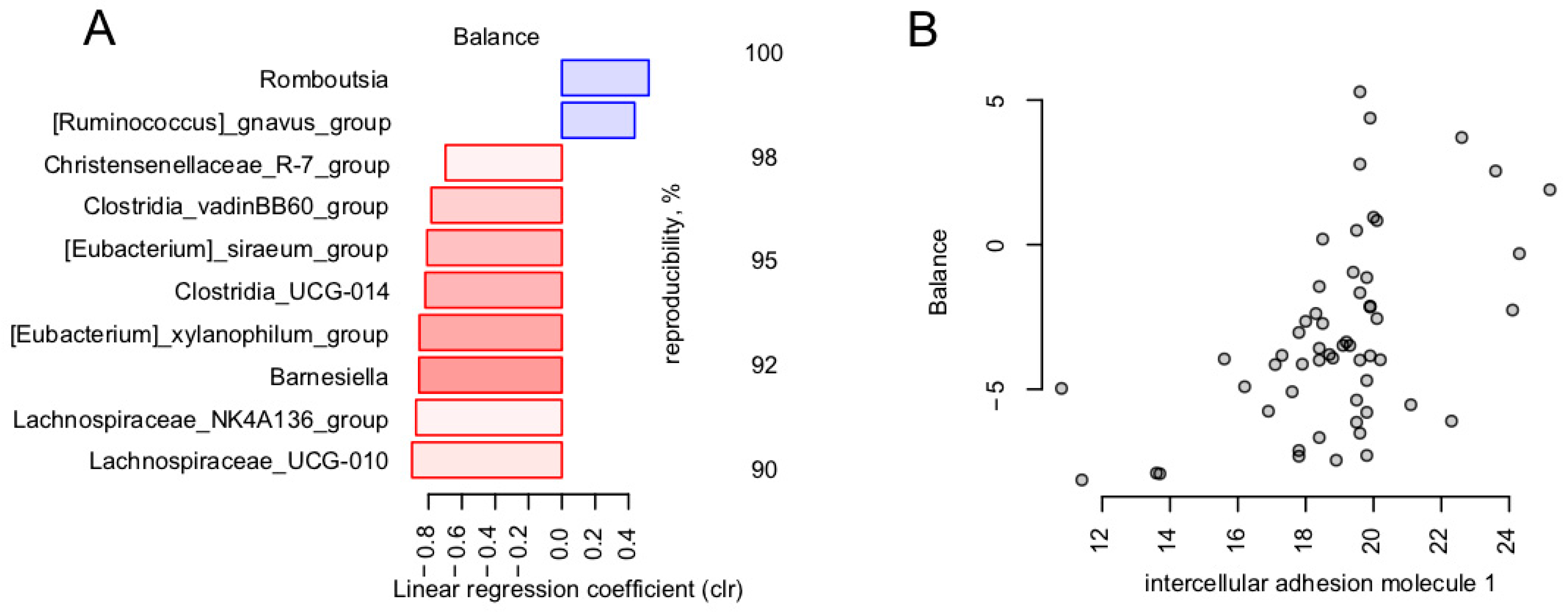
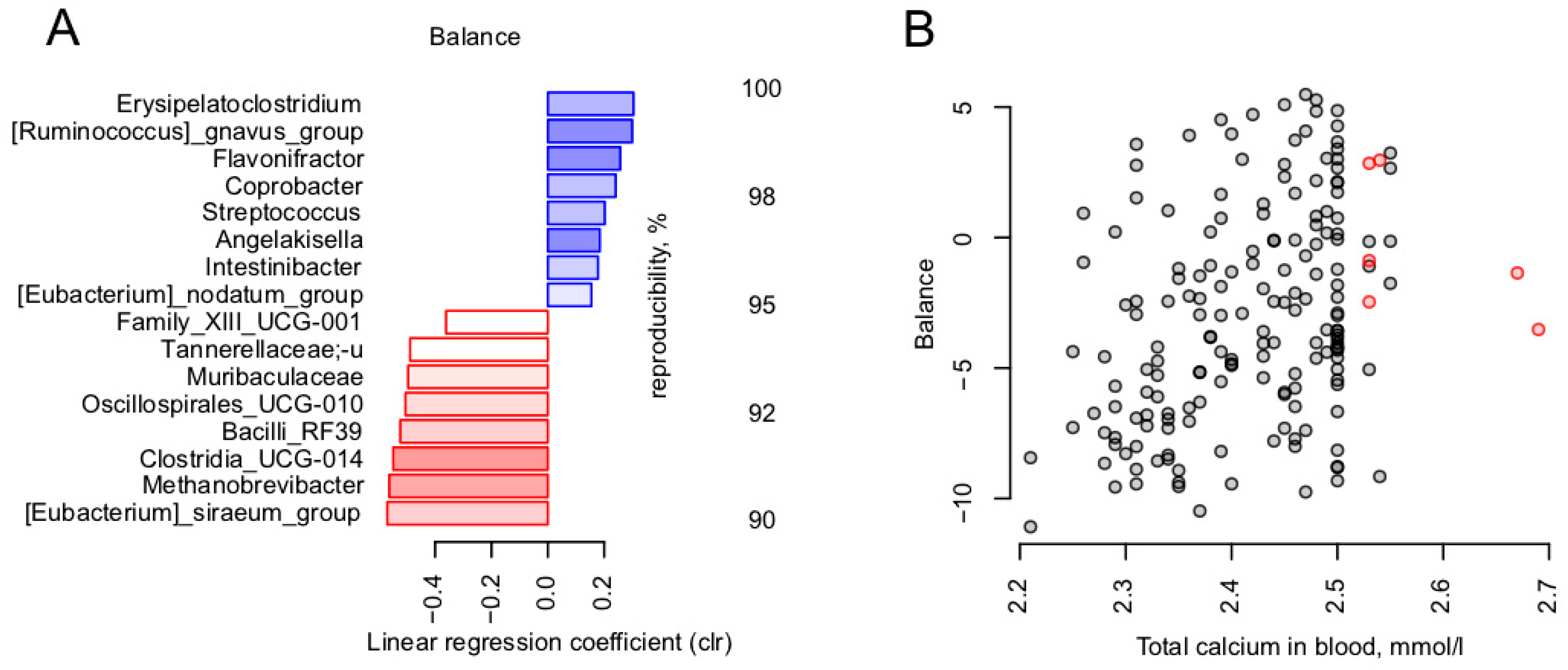
3.4. Calcium Is the Blood Metabolite Most Strongly Associated with Gut Microbiome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-Based Drug Repurposing for Novel Coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Guo, C.; Tang, L.; Hong, Z.; Zhou, J.; Dong, X.; Yin, H.; Xiao, Q.; Tang, Y.; Qu, X.; et al. Prolonged Presence of SARS-CoV-2 Viral RNA in Faecal Samples. Lancet Gastroenterol. Hepatol. 2020, 5, 434–435. [Google Scholar] [CrossRef]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA Expression Profiling of ACE 2, a Novel Homologue of Angiotensin Converting Enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.K.; To, K.-F.; Chan, P.K.S.; Chan, H.L.Y.; Wu, A.K.L.; Lee, N.; Yuen, K.Y.; Sung, J.J.Y. Enteric Involvement of Severe Acute Respiratory Syndrome-Associated Coronavirus Infection. Gastroenterology 2003, 125, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A Familial Cluster of Pneumonia Associated with the 2019 Novel Coronavirus Indicating Person-to-Person Transmission: A Study of a Family Cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Heneghan, C.J.; Spencer, E.A.; Brassey, J.; Plüddemann, A.; Onakpoya, I.J.; Evans, D.H.; Conly, J.M.; Jefferson, T. SARS-CoV-2 and the Role of Orofecal Transmission: A Systematic Review. F1000Res 2021, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Groves, H.T.; Higham, S.L.; Moffatt, M.F.; Cox, M.J.; Tregoning, J.S. Respiratory Viral Infection Alters the Gut Microbiota by Inducing Inappetence. MBio 2020, 11, 434–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, R.P. The Microbiome and Critical Illness. Lancet Respir. Med. 2016, 4, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, A.; Bernard, L.; Poquet, Y.; Lugo-Villarino, G.; Neyrolles, O. The Role of the Lung Microbiota and the Gut-Lung Axis in Respiratory Infectious Diseases. Cell. Microbiol. 2018, 20, e12966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front. Immunol. 2019, 10, 2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, D. Understanding Immune Variation for Improved Translational Medicine. Curr. Opin. Immunol. 2020, 65, 83–88. [Google Scholar] [CrossRef]
- Duffy, D. Milieu Intérieur: Defining the Boundaries of a Healthy Immune Response for Improved Vaccination Strategies. Hum. Vaccines Immunother. 2018, 14, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Sadrekarimi, H.; Gardanova, Z.R.; Bakhshesh, M.; Ebrahimzadeh, F.; Yaseri, A.F.; Thangavelu, L.; Hasanpoor, Z.; Zadeh, F.A.; Kahrizi, M.S. Emerging Role of Human Microbiome in Cancer Development and Response to Therapy: Special Focus on Intestinal Microflora. J. Transl. Med. 2022, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of Gut Microbiota in Type 2 Diabetes Pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Wilson Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome–Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef]
- Zalar, B.; Haslberger, A.; Peterlin, B. The Role of Microbiota in Depression-A Brief Review. Psychiatr. Danub. 2018, 30, 136–141. [Google Scholar] [CrossRef]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut Microbiome and Aging: Physiological and Mechanistic Insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Looft, T.; Allen, H.K. Collateral Effects of Antibiotics on Mammalian Gut Microbiomes. Gut Microbes 2012, 3, 463–467. [Google Scholar] [CrossRef]
- Ma, W.A.; Pfeiffer, J.K. Microbiota-Independent Antiviral Effects of Antibiotics on Poliovirus and Coxsackievirus. Virology 2020, 546, 20–24. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef]
- Gou, W.; Fu, Y.; Yue, L.; Chen, G.-D.; Cai, X.; Shuai, M.; Xu, F.; Yi, X.; Chen, H.; Zhu, Y.; et al. Gut Microbiota, Inflammation, and Molecular Signatures of Host Response to Infection. J. Genet. Genom. 2021, 48, 792–802. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Newsome, R.C.; Gauthier, J.; Hernandez, M.C.; Abraham, G.E.; Robinson, T.O.; Williams, H.B.; Sloan, M.; Owings, A.; Laird, H.; Christian, T.; et al. The Gut Microbiome of COVID-19 Recovered Patients Returns to Uninfected Status in a Minority-Dominated United States Cohort. Gut Microbes 2021, 13, 1926840. [Google Scholar] [CrossRef]
- Chen, Y.; Gu, S.; Chen, Y.; Lu, H.; Shi, D.; Guo, J.; Wu, W.-R.; Yang, Y.; Li, Y.; Xu, K.-J.; et al. Letter: Six-Month Follow-up of Gut Microbiota Richness in Patients with COVID-19. Gut 2022, 71, 222. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Mak, J.W.Y.; Su, Q.; Yeoh, Y.K.; Lui, G.C.-Y.; Ng, S.S.S.; Zhang, F.; Li, A.Y.L.; Lu, W.; Hui, D.S.-C.; et al. Gut Microbiota Dynamics in a Prospective Cohort of Patients with Post-Acute COVID-19 Syndrome. Gut 2022, 71, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An Improved Dual-Indexing Approach for Multiplexed 16S rRNA Gene Sequencing on the Illumina MiSeq Platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimova, D.; Tyakht, A.; Popenko, A.; Vasilyev, A.; Altukhov, I.; Dovidchenko, N.; Odintsova, V.; Klimenko, N.; Loshkarev, R.; Pashkova, M.; et al. Knomics-Biota-a System for Exploratory Analysis of Human Gut Microbiota Data. BioData Min. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2′s q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. DegePrime, a Program for Degenerate Primer Design for Broad-Taxonomic-Range PCR in Microbial Ecology Studies. Appl. Environ. Microbiol. 2014, 80, 16. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A mathematical theory of communication. IEEE 1948, 27, 379–423. [Google Scholar]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Aitchison, J. The Statistical Analysis of Compositional Data; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higginson, E.E.; Sayeed, M.A.; Pereira Dias, J.; Shetty, V.; Ballal, M.; Srivastava, S.K.; Willis, I.; Qadri, F.; Dougan, G.; Mutreja, A. Microbiome Profiling of Enterotoxigenic Escherichia Coli (ETEC) Carriers Highlights Signature Differences between Symptomatic and Asymptomatic Individuals. MBio 2022, 13, e0015722. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of Previously Uncultured Members of the Human Gut Microbiota by Culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Odintsova, V.E.; Klimenko, N.S.; Tyakht, A.V. Approximation of a Microbiome Composition Shift by a Change in a Single Balance Between Two Groups of Taxa. mSystems 2022, 28, e0015522. [Google Scholar] [CrossRef] [PubMed]
- Tyakht, A.V.; Kostryukova, E.S.; Popenko, A.S.; Belenikin, M.S.; Pavlenko, A.V.; Larin, A.K.; Karpova, I.Y.; Selezneva, O.V.; Semashko, T.A.; Ospanova, E.A.; et al. Human Gut Microbiota Community Structures in Urban and Rural Populations in Russia. Nat. Commun. 2013, 4, 2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.L.; Chung, A.C.K.; Cheung, C.P.; Tso, E.Y.K.; Fung, K.S.C.; et al. Gut Microbiota Composition Reflects Disease Severity and Dysfunctional Immune Responses in Patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.; Zhang, D.; Ma, W.-L.; Wang, X. Linking the Gut Microbiota to Persistent Symptoms in Survivors of COVID-19 after Discharge. J. Microbiol. 2021, 59, 941–948. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, Y.; Zuo, T.; Yeoh, Y.K.; Liu, Q.; Zhang, L.; Zhan, H.; Lu, W.; Xu, W.; Lui, G.C.Y.; et al. Prolonged Impairment of Short-Chain Fatty Acid and L-Isoleucine Biosynthesis in Gut Microbiome in Patients With COVID-19. Gastroenterology 2022, 162, 548–561.e4. [Google Scholar] [CrossRef]
- Buonacera, A.; Stancanelli, B.; Colaci, M.; Malatino, L. Neutrophil to Lymphocyte Ratio: An Emerging Marker of the Relationships between the Immune System and Diseases. Int. J. Mol. Sci. 2022, 23, 3636. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Y.; Wang, X.; Yang, R.; Zhu, X.; Zhang, Y.; Chen, C.; Yuan, H.; Yang, Z.; Sun, L. Gut Bacteria Akkermansia Is Associated with Reduced Risk of Obesity: Evidence from the American Gut Project. Nutr. Metab. 2020, 17, 1–9. [Google Scholar] [CrossRef]
- Waters, J.L.; Ley, R.E. The Human Gut Bacteria Christensenellaceae Are Widespread, Heritable, and Associated with Health. BMC Biol. 2019, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus Gnavus, a Member of the Human Gut Microbiome Associated with Crohn’s Disease, Produces an Inflammatory Polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [Green Version]
- Toya, T.; Corban, M.T.; Marrietta, E.; Horwath, I.E.; Lerman, L.O.; Murray, J.A.; Lerman, A. Coronary Artery Disease Is Associated with an Altered Gut Microbiome Composition. PLoS ONE 2020, 15, e0227147. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Wu, H.; Wu, S.-D.; Lu, N.; Wang, Y.-T.; Liu, H.-N.; Dong, L.; Liu, T.-T.; Shen, X.-Z. Parasutterella, in Association with Irritable Bowel Syndrome and Intestinal Chronic Inflammation. J. Gastroenterol. Hepatol. 2018, 33, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Ogita, T.; Yamamoto, Y.; Mikami, A.; Shigemori, S.; Sato, T.; Shimosato, T. Oral Administration of Flavonifractor Plautii Strongly Suppresses Th2 Immune Responses in Mice. Front. Immunol. 2020, 11, 379. [Google Scholar] [CrossRef] [Green Version]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond Gut Feelings: How the Gut Microbiota Regulates Blood Pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef]
- Astudillo, A.A.; Mayrovitz, H.N. The Gut Microbiome and Cardiovascular Disease. Cureus 2021, 16, e14519. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and Disease Markers Correlate with Gut Microbiome Composition across Thousands of People. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut Microbiota Dysbiosis Contributes to the Development of Hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Al Khodor, S.; Reichert, B.; Shatat, I.F. The Microbiome and Blood Pressure: Can Microbes Regulate Our Blood Pressure? Front. Pediatr. 2017, 5, 138. [Google Scholar] [CrossRef] [Green Version]
- Kashtanova, D.; Tkacheva, O.; Popenko, A.; Egshatyan, L.; Tyakht, A.; Alexeev, D.; Kotovskaya, Y.; Plokhova, E.; Boytsov, S. Gut Microbiota and Vascular Biomarkers in Patients without Clinical Cardiovascular Diseases. Artery Res. 2017, 18, 41–48. [Google Scholar] [CrossRef]
- Larsen, J.M. The Immune Response to Prevotella Bacteria in Chronic Inflammatory Disease. Immunology 2017, 151, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, X.; Liu, X.; Chen, D.; Wang, M.; Jiang, X.; Xiong, Z. The Roles of the Gut Microbiota and Chronic Low-Grade Inflammation in Older Adults With Frailty. Front. Cell. Infect. Microbiol. 2021, 1, 675414. [Google Scholar] [CrossRef] [PubMed]
- van den Munckhof, I.C.L.; Kurilshikov, A.; Ter Horst, R.; Riksen, N.P.; Joosten, L.A.B.; Zhernakova, A.; Fu, J.; Keating, S.T.; Netea, M.G.; de Graaf, J.; et al. Role of Gut Microbiota in Chronic Low-Grade Inflammation as Potential Driver for Atherosclerotic Cardiovascular Disease: A Systematic Review of Human Studies. Obes. Rev. 2018, 19, 1719–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, T.; de Hase, E.M.; Van Hul, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Depommier, C.; Druart, C.; Everard, A.; Maiter, D.; et al. Dysosmobacter Welbionis Is a Newly Isolated Human Commensal Bacterium Preventing Diet-Induced Obesity and Metabolic Disorders in Mice. Gut 2022, 71, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Lagier, J.C.; Yahav, D.; Paul, M. Gut Bacterial Microbiota and Obesity. Clin. Microbiol. Infect. 2013, 19, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharmacol. 2018, 25, 1082. [Google Scholar] [CrossRef]
- Lurie-Weinberger, M.N.; Gophna, U. Archaea in and on the Human Body: Health Implications and Future Directions. PLoS Pathog. 2015, 11, e1004833. [Google Scholar] [CrossRef]
- Shadi Ahmadmehrabi, W.H.W.T. Gut Microbiome and Its Role in Cardiovascular Diseases. Curr. Opin. Cardiol. 2017, 32, 761. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Atay, A.E.; Esen, B.; Akbas, H.; Gokmen, E.S.; Pilten, S.; Guler, H.; Yavuz, D.G. Serum ICAM-1 level and ICAM-1 gene 1462A>G (K469E) polimorphism on microalbuminuria in nondiabetic, nonhypertensive and normolipidemic obese patients: Genetical background of microalbuminuria in obesity. Nefrología 2017, 37, 381–388. [Google Scholar] [CrossRef]
- Mandić, A.D.; Woting, A.; Jaenicke, T.; Sander, A.; Sabrowski, W.; Rolle-Kampcyk, U.; von Bergen, M.; Blaut, M. Clostridium Ramosum Regulates Enterochromaffin Cell Development and Serotonin Release. Sci. Rep. 2019, 9, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, L.; Raj, R.; Rhoad, W.; Akella, R.; Jacob, A. Immobilization-Induced Hypercalcemia in COVID-19 With a Prolonged Intensive Care Unit Stay. Cureus 2022, 14, e24081. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Units | Group A (Med, Q1; Q3) | Group N (Med, Q1; Q3) | Group S (Med, Q1; Q3) |
|---|---|---|---|---|
| Ultrasound dopplerography of the carotid arteries | ||||
| Intima media thickness right | cm | 0.73; 0.64, 0.84 (n = 46) | 0.73; 0.64, 0.88 (n = 48) | 0.88; 0.74, 0.98 (n = 86) |
| Maximal stenosis | 0; 0, 30 (n = 46) | 0; 0, 25 (n = 48) | 25; 0, 35 (n = 86) | |
| ECHO-CG | ||||
| Myocardial mass index | g/m2 | 62.5; 55, 73.83 (n = 42) | 64.5; 54.25, 74 (n = 46) | 75; 62.25, 88.75 (n = 74) |
| Anteroposterior dimension of the right ventricle | cm | 2.9; 2.7, 3.1 (n = 46) | 2.8; 2.6, 3.1 (n = 45) | 3.1; 2.8, 3.4 (n = 85) |
| Left atrial volume | mL | 44.5; 38.25, 54.75 (n = 46) | 48; 36.5, 57 (n = 47) | 56; 45.5, 68.5 (n = 83) |
| Right atrial volume | mL | 33.5; 28, 39.5 (n = 46) | 32; 27, 40 (n = 47) | 36; 32.25, 43.75 (n = 82) |
| Systolic pressure in the pulmonary artery | mmHg | 25; 24, 27 (n = 46) | 25; 24, 30 (n = 47) | 27; 25, 30 (n = 85) |
| E/A ratio | 1.27; 0.85, 1.6 (n = 46) | 1.3; 0.97, 1.5 (n = 47) | 0.8; 0.7, 1 (n = 81) | |
| E/e’ ratio | 6.5; 5, 7 (n = 46) | 7; 6, 8 (n = 47) | 8; 7, 10 (n = 80) | |
| Left ventricular diastolic dysfunction | 2; 1, 2 (n = 46) | 2; 1, 2 (n = 47) | 2; 2, 2 (n = 82) | |
| End diastolic volume | mL | 92; 82.75, 107.5 (n = 46) | 88; 71.25, 109.5 (n = 46) | 108; 92, 118 (n = 85) |
| Ejection fraction | 60.5; 60, 64 (n = 46) | 63; 60, 64.5 (n = 47) | 60; 58, 62 (n = 85) | |
| Applanation tonometry | ||||
| Pulse Wave Velocity | m/s | 9.7; 7.9, 10.67 (n = 46) | 8.6; 6.8, 10.3 (n = 44) | 10.5; 9.1, 12.1 (n = 85) |
| Central systolic blood pressure | mmHg | 117; 108.25, 124.75 (n = 46) | 113.5; 106.25, 123.5 (n = 46) | 125; 115.25, 134 (n = 86) |
| Central diastolic blood pressure | mmHg | 79.5; 71.25, 84.75 (n = 46) | 76; 71, 81.75 (n = 46) | 86; 79.25, 91 (n = 86) |
| Augmentation Index | 25; 14, 29 (n = 46) | 21; 13.25, 30.25 (n = 46) | 24.5; 17.25, 30 (n = 86) | |
| Sitting heart rate | bpm | 71; 64, 76 (n = 46) | 68; 65, 77 (n = 48) | 72; 66, 78 (n = 86) |
| Sitting pulse blood pressure | mmHg | 45.5; 41.25, 52.75 (n = 46) | 45; 40, 50 (n = 48) | 50; 44, 60 (n = 86) |
| Concentric remodeling | Yes|No | 10|36 (n = 46) | 19|28 (n = 47) | 32|53 (n = 85) |
| Degree of mitral regurgitation | 1st|2nd | 37|8 (n = 45) | 41|6 (n = 47) | 50|34 (n = 84) |
| Cooperative Used as a Balance Numerator | Factor | Linear Model Coefficient | p | FDR | Number of Subjects |
|---|---|---|---|---|---|
| Lactobacillus-coop | Pulse pressure in sitting position (PP) | 0.8576 | 0 | 0.0034 | 180 |
| Lactobacillus-coop | Central systolic blood pressure (CSBP) | 0.6591 | 0.0009 | 0.0468 | 178 |
| Prevotellaceae-coop | Pulse Wave Velocity (PWV) | 0.6488 | 0.0003 | 0.0357 | 175 |
| Prevotellaceae-coop | Pulse pressure in sitting position (PP) | 0.5846 | 0.001 | 0.0468 | 180 |
| Subdoligranulum-coop | Pulse pressure in sitting position (PP) | −0.4016 | 0.0018 | 0.0703 | 180 |
| Subdoligranulum-coop | Maximum percentage of stenosis (MPS) | −0.427 | 0.0009 | 0.0468 | 180 |
| Subdoligranulum-coop | Systolic pulmonary artery pressure (sPAP) (highly correlated with: tricuspid regurgitation gradient) * | −0.4658 | 0.0002 | 0.0351 | 178 |
| Methanobrevibacter-coop | End-diastolic volume (EDV) (highly correlated with: end-diastolic size) * | −0.6481 | 0.0004 | 0.0357 | 177 |
| Parameter | Group N (Med, Q1; Q3) | Group A (Med, Q1; Q3) | Group S (Med, Q1; Q3) |
|---|---|---|---|
| VEGF-R1 | 0.2; 0.14, 0.26 (n = 42) | 0.18; 0.14, 0.25 (n = 47) | 0.19; 0.13, 0.28 (n = 79) |
| ICAM-1 | 19.6; 17.8, 19.8 (n = 9) | 18.2; 17.65, 19.75 (n = 14) | 19.5; 18.5, 19.9 (n = 33) |
| VCAM-1 | 12.65; 8.54, 27.3 (n = 42) | 11.4; 8.21, 31.8 (n = 47) | 19.7; 10.15, 38.45 (n = 79) |
| E-selectin | 7.32; 5.8, 11.38 (n = 42) | 8.1; 5.08, 12.4 (n = 47) | 8.38; 6.27, 12.6 (n = 79) |
| von Willebrand factor, % | 103.5; 85.5, 122.5 (n = 46) | 116; 93, 145 (n = 46) | 143; 117, 177 (n = 85) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkacheva, O.N.; Klimenko, N.S.; Kashtanova, D.A.; Tyakht, A.V.; Maytesyan, L.V.; Akopyan, A.A.; Koshechkin, S.I.; Strazhesko, I.D. Gut Microbiome in Post-COVID-19 Patients Is Linked to Immune and Cardiovascular Health Status but Not COVID-19 Severity. Microorganisms 2023, 11, 1036. https://doi.org/10.3390/microorganisms11041036
Tkacheva ON, Klimenko NS, Kashtanova DA, Tyakht AV, Maytesyan LV, Akopyan AA, Koshechkin SI, Strazhesko ID. Gut Microbiome in Post-COVID-19 Patients Is Linked to Immune and Cardiovascular Health Status but Not COVID-19 Severity. Microorganisms. 2023; 11(4):1036. https://doi.org/10.3390/microorganisms11041036
Chicago/Turabian StyleTkacheva, Olga N., Natalia S. Klimenko, Daria A. Kashtanova, Alexander V. Tyakht, Lilit V. Maytesyan, Anna A. Akopyan, Stanislav I. Koshechkin, and Irina D. Strazhesko. 2023. "Gut Microbiome in Post-COVID-19 Patients Is Linked to Immune and Cardiovascular Health Status but Not COVID-19 Severity" Microorganisms 11, no. 4: 1036. https://doi.org/10.3390/microorganisms11041036