

A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
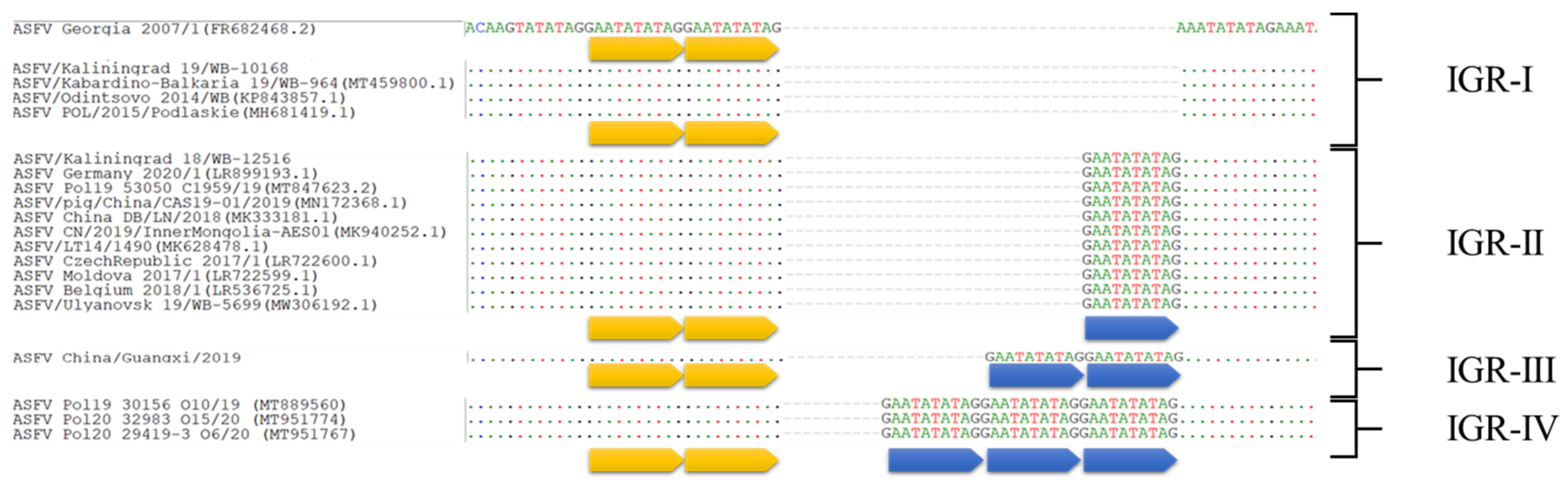{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Essential Genetic Markers Used for Subtyping Genotype II ASFVs
- 1. B646L : Genotyping marker.
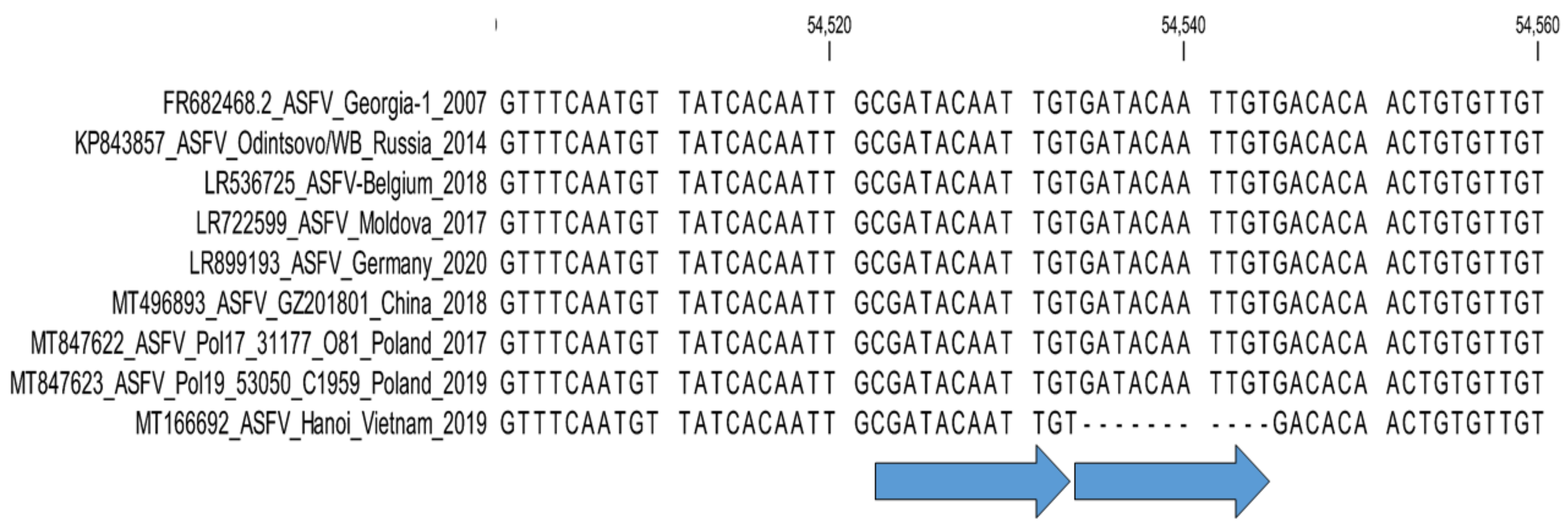
- 2. Central variable region (CVR) of B602L gene: Sub-genotyping marker.
- 3. Intergenic region I73R/I329L: Sub-genotyping marker.
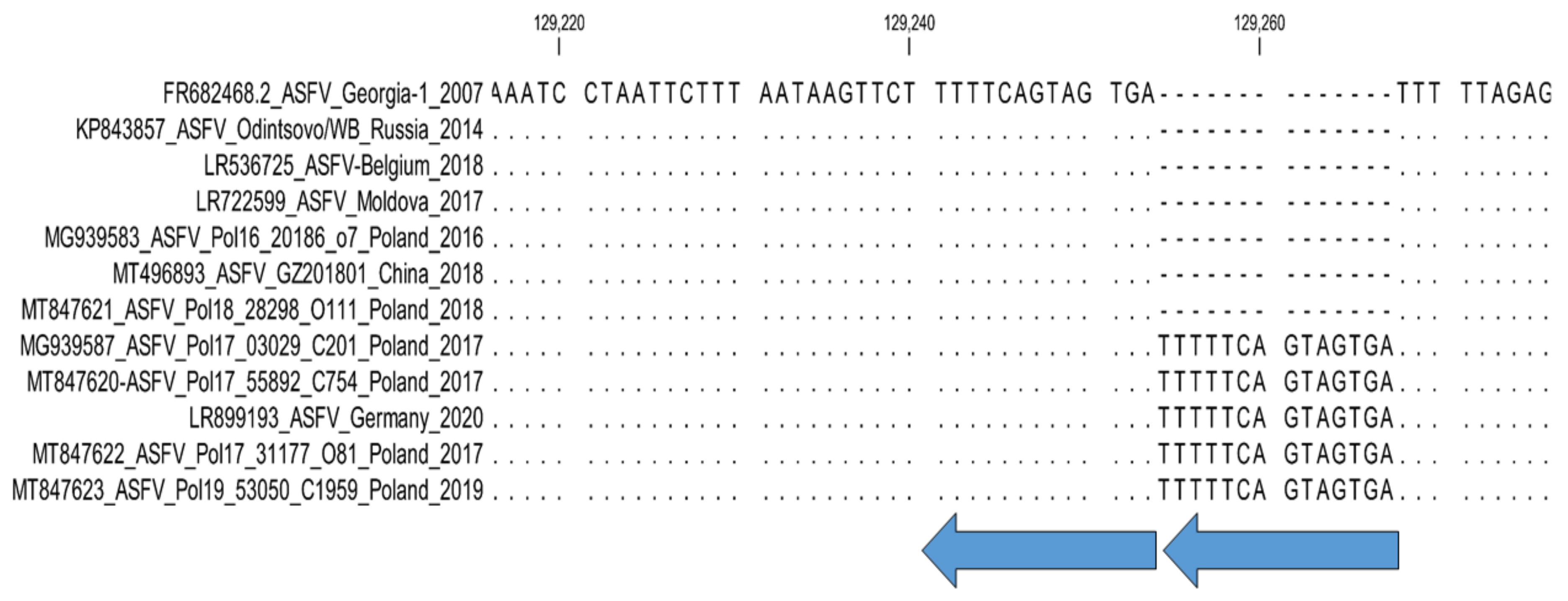
- 4. Intergenic region MGF-505-9R/10R: Sub-genotyping marker.
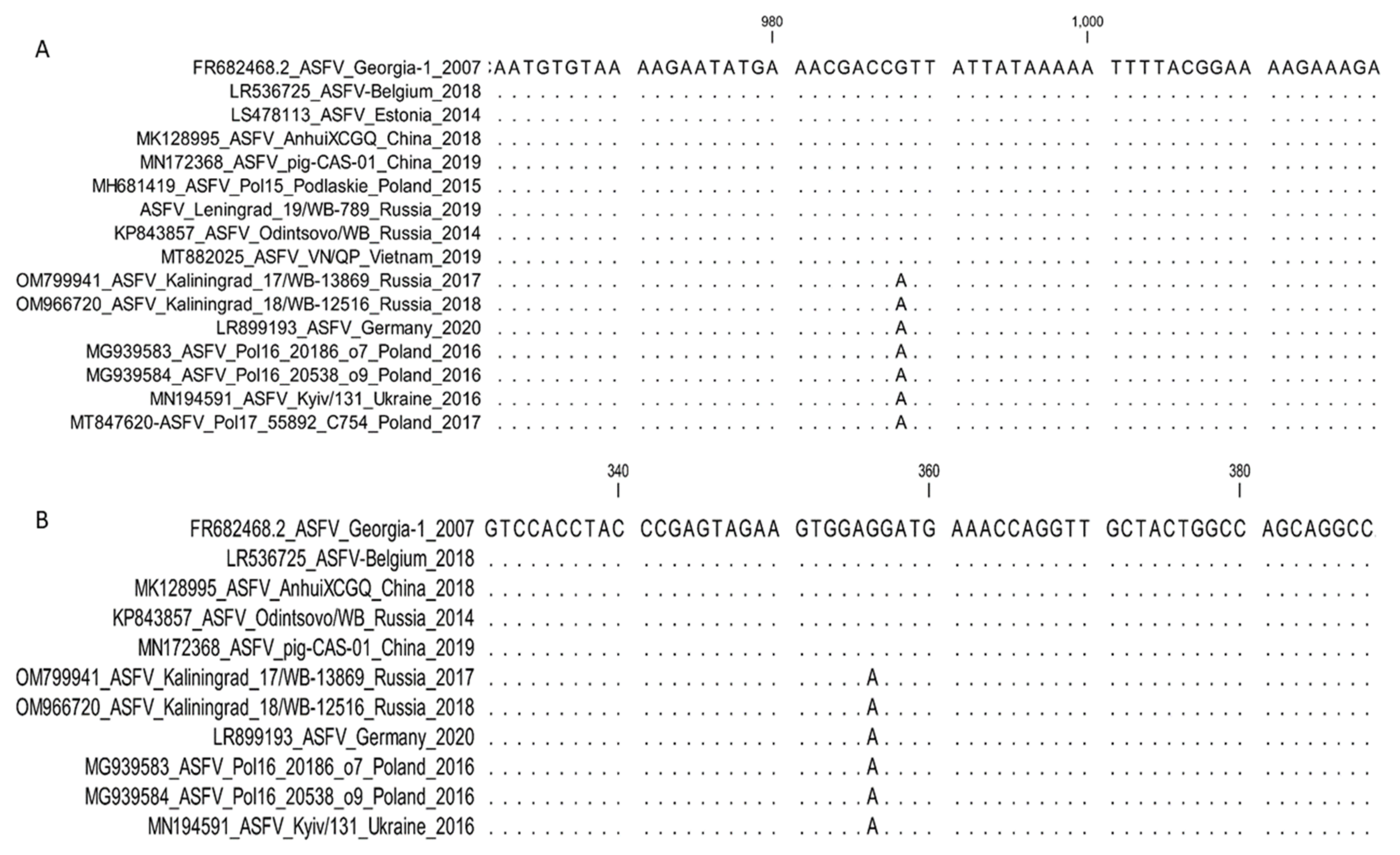
- 5. K145R: Sub-genotyping marker.
- 6. O174L : Sub-genotyping marker.
3. Additional Sub-Genotyping Markers
- 1. Intergenic region A179L/A137R: Sub-genotyping marker.
- 2. MGF-505-5R and MGF-110-7L: Sub-genotyping markers.
- 3. I267L: Sub-genotyping marker.
- 4. MGF-360-10L and MGF-505-9R: Sub-genotyping markers.
- 5. Intergenic region C315R/C147L: Sub-genotyping marker.
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Galindo, I.; Alonso, C. African swine fever virus: A review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Denyer, M.S.; Wilkinson, P.J. African Swine Fever. In Encyclopedia of Immunology; Academic Press: Cambridge, MA, USA, 1998; pp. 54–56. [Google Scholar]
- Yanez, R.J.; Rodríguez, J.M.; Nogal, M.L.; Yuste, L.; Enríquez, C.; Rodriguez, J.F.; Vinuela, E. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.G.; R Thomson, G. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Quembo, C.J.; Jori, F.; Vosloo, W.H.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.E. On a form of swine fever occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 243–262. [Google Scholar]
- Alvarez, A.O.; Marcotegui, M.A. African swine fever-clinical aspects. In African Swine Fever; Springer: Boston, MA, USA, 1987; pp. 11–20. [Google Scholar]
- Franzoni, G.; Dei Giudici, S.; Loi, F.; Sanna, D.; Floris, M.; Fiori, M.; Sanna, M.L.; Madrau, P.; Scarpa, F.; Zinellu, S.; et al. African swine fever circulation among free-ranging pigs in Sardinia: Data from the eradication program. Vaccines 2020, 8, 549. [Google Scholar] [CrossRef]
- Beltrán-Alcrudo, D.; Lubroth, J.; Depner, K.; De La Rocque, S. African swine fever in the Caucasus. FAO Empres Watch 2008, 1, 1–8. [Google Scholar]
- OIE-WAHIS. African Swine Fever (ASF)–Situation Report 3 of 12 January 2022. Available online: https://www.woah.org/app/uploads/2022/01/asf-situation-report-3.pdf (accessed on 22 January 2023).
- Gonzales, W.; Moreno, C.; Duran, U.; Henao, N.; Bencosme, M.; Lora, P.; Reyes, R.; Núñez, R.; De Gracia, A.; Perez, A.M. African swine fever in the Dominican Republic. Transbound. Emerg. Dis. 2021, 68, 3018–3019. [Google Scholar] [CrossRef]
- Sun, E.; Huang, L.; Zhang, X.; Zhang, J.; Shen, D.; Zhang, Z.; Wang, Z.; Huo, H.; Wang, W.; Huangfu, H.; et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg. Microbes Infect. 2021, 10, 2183–2193. [Google Scholar] [CrossRef]
- Malogolovkin, A.; Yelsukova, A.; Gallardo, C.; Tsybanov, S.; Kolbasov, D. Molecular characterization of African swine fever virus isolates originating from outbreaks in the Russian Federation between 2007 and 2011. Vet. Microbiol. 2012, 158, 415–419. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Shotin, A.; Igolkin, A.; Shevchenko, I.; Gruzdev, K.N.; Vlasova, N. Comparative analysis of full genome sequences of African swine fever virus isolates taken from wild boars in Russia in 2019. Pathogens 2021, 10, 521. [Google Scholar] [CrossRef]
- Wesley, R.D.; Tuthill, A.E. Genome relatedness among African swine fever virus field isolates by restriction endonuclease analysis. Prev. Vet. Med. 1984, 2, 53–62. [Google Scholar] [CrossRef]
- Blasco, R.; Agüero, M.; Almendral, J.; Vinuela, E. Variable and constant regions in African swine fever virus DNA. Virology 1989, 168, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Wilkinson, P.J. Genetic diversity of African swine fever virus isolates from soft ticks (Ornithodoros moubata) inhabiting warthog burrows in Zambia. J. Gen. Virol. 1988, 69, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a new genotype of African swine fever virus in domestic pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef]
- Alkhamis, M.A.; Gallardo, C.; Jurado, C.; Soler, A.; Arias, M.; Sanchez-Vizcaıno, J.M. Phylodynamics and evolutionary epidemiology of African swine fever p72-CVR genes in Eurasia and Africa. PLoS ONE 2018, 13, e0192565. [Google Scholar] [CrossRef] [Green Version]
- Irusta, P.M.; Borca, M.V.; Kutish, G.F.; Lu, Z.; Caler, E.; Carrillo, C.; Rock, D.L. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology 1996, 220, 20–27. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.; Gerber, L.J.; Vosloo, W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Phologane, S.B.; Bastos, A.D.; Penrith, M.L. Intra-and inter-genotypic size variation in the central variable region of the 9RL open reading frame of diverse African swine fever viruses. Virus Genes 2005, 31, 357–360. [Google Scholar] [CrossRef]
- Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular characterization of African swine fever virus isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Shotin, A.; Korennoy, F.I.; Igolkin, A.; Sprygin, A. Genetic Characterization of the Central Variable Region in African Swine Fever Virus Isolates in the Russian Federation from 2013 to 2017. Pathogens 2022, 11, 919. [Google Scholar] [CrossRef]
- Kim, H.J.; Cho, K.H.; Ryu, J.H.; Jang, M.K.; Chae, H.G.; Choi, J.D.; Nah, J.J.; Kim, Y.J.; Kang, H.E. Isolation and genetic characterization of African swine fever virus from domestic pig farms in South Korea, 2019. Viruses 2020, 12, 1237. [Google Scholar] [CrossRef] [PubMed]
- Mai, N.T.; Vu, X.D.; Nguyen, T.T.; Nguyen, V.T.; Trinh, T.B.; Kim, Y.J.; Kim, H.J.; Cho, K.H.; Nguyen, T.L.; Bui, T.T.; et al. Molecular profile of African swine fever virus (ASFV) circulating in Vietnam during 2019–2020 outbreaks. Arch. Virol. 2021, 166, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Liu, H.; Si, H.; Long, F.; Feng, S. Molecular characterization of African swine fever virus from 2019–2020 outbreaks in Guangxi province, southern China. Front. Vet. Sci. 2022, 9, 912224. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Perez, C.; Simon, A.; Ivanova, E.; Donescu, D.; et al. A multi gene-approach genotyping method identifies twenty-four genetic clusters within the genotype II-European African swine fever viruses (ASFVs) circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef] [PubMed]
- Mazloum, A.; Igolkin, A.S.; Vlasova, N.N.; Romenskaya, D.V. African swine fever virus: Use of genetic markers in analysis of its routes of spread. Vet. Sci. Today 2019, 2, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg. Infect. Dis. 2014, 20, 1544. [Google Scholar] [CrossRef] [Green Version]
- Elsukova, A.; Shevchenko, I.; Varentsova, A.; Puzankova, O.; Zhukov, I.Y.; Pershin, A.S. Biological properties of African swine fever virus Odintsovo 02/14 isolate and its genome analysis. Int. J. Environ. Agricult. Res. 2017, 3, 26–37. [Google Scholar]
- Ge, S.; Liu, Y.; Li, L.; Wang, Q.; Li, J.; Ren, W.; Liu, C.; Bao, J.; Wu, X.; Wang, Z. An extra insertion of tandem repeat sequence in African swine fever virus, China, 2019. Virus Genes 2019, 55, 843–847. [Google Scholar] [CrossRef]
- Tran, H.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Nguyen, C.T.; Chu, N.T.; Hoang, T.V.; Nguyen, H.T.; Dang, H.V. Circulation of two different variants of intergenic region (IGR) located between the I73R and I329L genes of African swine fever virus strains in Vietnam. Transbound. Emerg. Dis. 2021, 68, 2693–2695. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Cho, K.H.; Mai, N.T.; Park, J.Y.; Trinh, T.B.; Jang, M.K.; Nguyen, T.T.; Vu, X.D.; Nguyen, T.L.; Nguyen, V.D.; et al. Multiple variants of African swine fever virus circulating in Vietnam. Arch. Virol. 2022, 167, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Hien, N.D.; Nguyen, L.T.; Hoang, L.T.; Bich, N.N.; Quyen, T.M.; Isoda, N.; Sakoda, Y. First Report of a Complete Genome Sequence of a Variant African Swine Fever Virus in the Mekong Delta, Vietnam. Pathogens 2022, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The spillover of African swine fever in Western Poland revealed its estimated origin on the basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L genomic sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Mazloum, A.; van Schalkwyk, A.; Shotin, A.; Zinyakov, N.; Igolkin, A.; Chernyshev, R.; Debeljak, Z.; Korennoy, F.; Sprygin, A.V. Whole-genome sequencing of African swine fever virus from wild boars in the Kaliningrad region reveals unique and distinguishing genomic mutations. Front. Vet. Sci. 2023, 9, 1019808. [Google Scholar] [CrossRef] [PubMed]
- Elsukova, A.; Shevchenko, I.; Varentsova, A.; Zinyakov, N.; Igolkin, A.; Vlasova, N. African swine fever (ASF), intergenic region, 9R/10R, NGS, tandem repeat sequences in the intergenic region MGF 505 9R/10R is a new marker of the genetic variability among ASF Genotype II viruses. In Proceedings of the EPIZONE, 10th Annual Meeting 2016, Madrid, Spain, 27–29 April 2016. [Google Scholar]
- Tran, H.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Chu, N.T.; Van Hoang, T.; Nguyen, H.T.; Netherton, C.L.; Dang, H.V. Novel method for sub-grouping of genotype II African swine fever viruses based on the intergenic region between the A179L and A137R genes. Vet. Med. Sci. 2022, 8, 607–609. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Woźniakowski, G.; Niemczuk, K. The first complete genomic sequences of African swine fever virus isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef] [Green Version]
- Mazur-Panasiuk, N.; Woźniakowski, G. The unique genetic variation within the O174L gene of Polish strains of African swine fever virus facilitates tracking virus origin. Arch. Virol. 2019, 164, 1667–1672. [Google Scholar] [CrossRef] [Green Version]
- Forth, J.H.; Calvelage, S.; Fischer, M.; Hellert, J.; Sehl-Ewert, J.; Roszyk, H.; Deutschmann, P.; Reichold, A.; Lange, M.; Thulke, H.H.; et al. African swine fever virus–variants on the rise. Emerg. Microbes Infect. 2023, 12, 2146537. [Google Scholar] [CrossRef]
- Wen, X.; He, X.; Zhang, X.; Zhang, X.; Liu, L.; Guan, Y.; Zhang, Y.; Bu, Z. Genome sequences derived from pig and dried blood pig feed samples provide important insights into the transmission of African swine fever virus in China in 2018. Emerg. Microbes Infect. 2019, 8, 303–306. [Google Scholar] [CrossRef]
- Mazloum, A.; Igolkin, A.S.; Shotin, A.R.; Zinyakov, N.G.; Vlasova, N.N.; Aronova, E.V.; Puzankova, O.S.; Gavrilova, V.L.; Shevchenko, I.V. Analysis of the whole-genome sequence of an ASF virus (Asfarviridae: Asfivirus: African swine fever virus) isolated from a wild boar (Sus scrofa) at the border between Russian Federation and Mongolia. Probl. Virol. 2022, 67, 153–164. [Google Scholar] [CrossRef]
- Chernyshev, R.; Sprygin, A.V.; Shotin, A.R.; Igolkin, A.; Mazloum, A. Comparative analysis of full genome sequences of African swine fever virus isolates taken from domestic pigs and wild boar in Zabaykalsky Krai of Russian Federation in 2020. Vet. Zootekhniya Biotekhnol. 2022, 10, 84–97. [Google Scholar] [CrossRef]
- Chernyshev, R.; Igolkin, A.; Sprygin, A.V.; Shotin, A.R.; Mazloum, A.; Chvala, I.A. Whole genome sequence analysis of the isolate ASFV/Primorsky-2019/WB-8235 of the African swine fever virus. Bull. Vet. Pharmacol. 2022, 4, 187–200. [Google Scholar] [CrossRef]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvili, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Shi, K.; Zhou, Q.; Xiong, C.; Mo, S.; Zhou, H.; Long, F.; Wei, H.; Hu, L.; Mo, M. The Development of a Multiplex Real-Time Quantitative PCR Assay for the Differential Detection of the Wild-Type Strain and the MGF505-2R, EP402R and I177L Gene-Deleted Strain of the African Swine Fever Virus. Animals 2022, 12, 1754. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Igolkin, A.; Heath, L.; Sprygin, A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 2023, 11, 642. https://doi.org/10.3390/microorganisms11030642
Mazloum A, van Schalkwyk A, Chernyshev R, Igolkin A, Heath L, Sprygin A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms. 2023; 11(3):642. https://doi.org/10.3390/microorganisms11030642
Chicago/Turabian StyleMazloum, Ali, Antoinette van Schalkwyk, Roman Chernyshev, Alexey Igolkin, Livio Heath, and Alexander Sprygin. 2023. "A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers" Microorganisms 11, no. 3: 642. https://doi.org/10.3390/microorganisms11030642