
Molecular Interactions of the Copper Chaperone Atx1 of Paracoccidioides brasiliensis with Fungal Proteins Suggest a Crosstalk between Iron and Copper Homeostasis

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Method
2.1. Strain Used and Culture Media
2.2. Obtaining Protein Extracts
2.3. Pull-Down Assays
2.4. Sample Preparation for nanoUPLC-MSE
2.5. High Performance Liquid Chromatography at Nanoscale Coupled to Mass Spectrometry
2.6. Spectra Processing and Proteomic Analysis
2.7. Recombinant Proteins and Polyclonal Antibodies
2.8. SDS-PAGE and Western Blotting
2.9. STRING Database Analysis
2.10. Prediction of PbAtx1 and PbCyb5 Structures
2.11. Molecular Docking
2.12. Far-Western Blot Analyses
3. Results
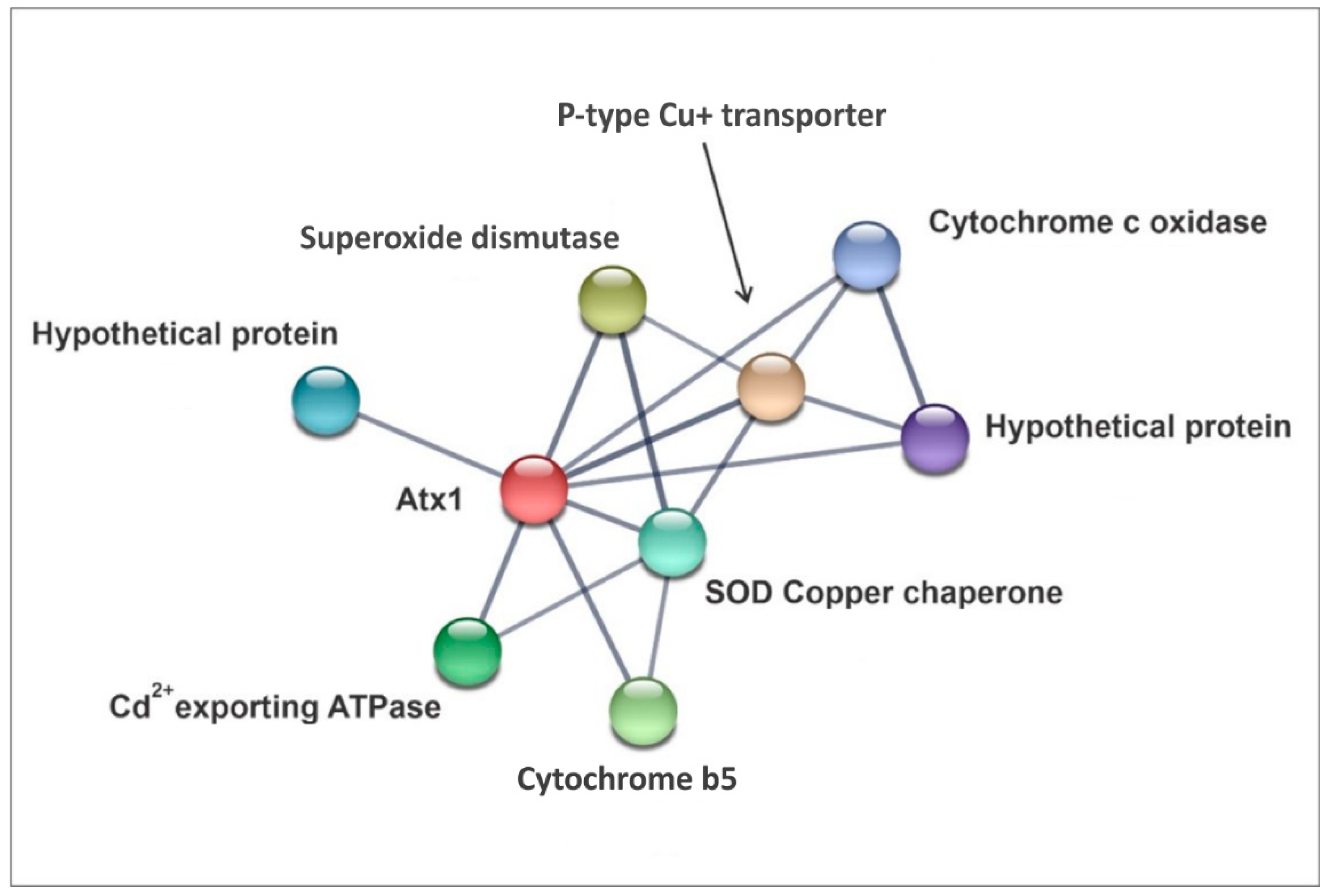
3.1. Overview of PbAtx1 Protein–Protein Interactions
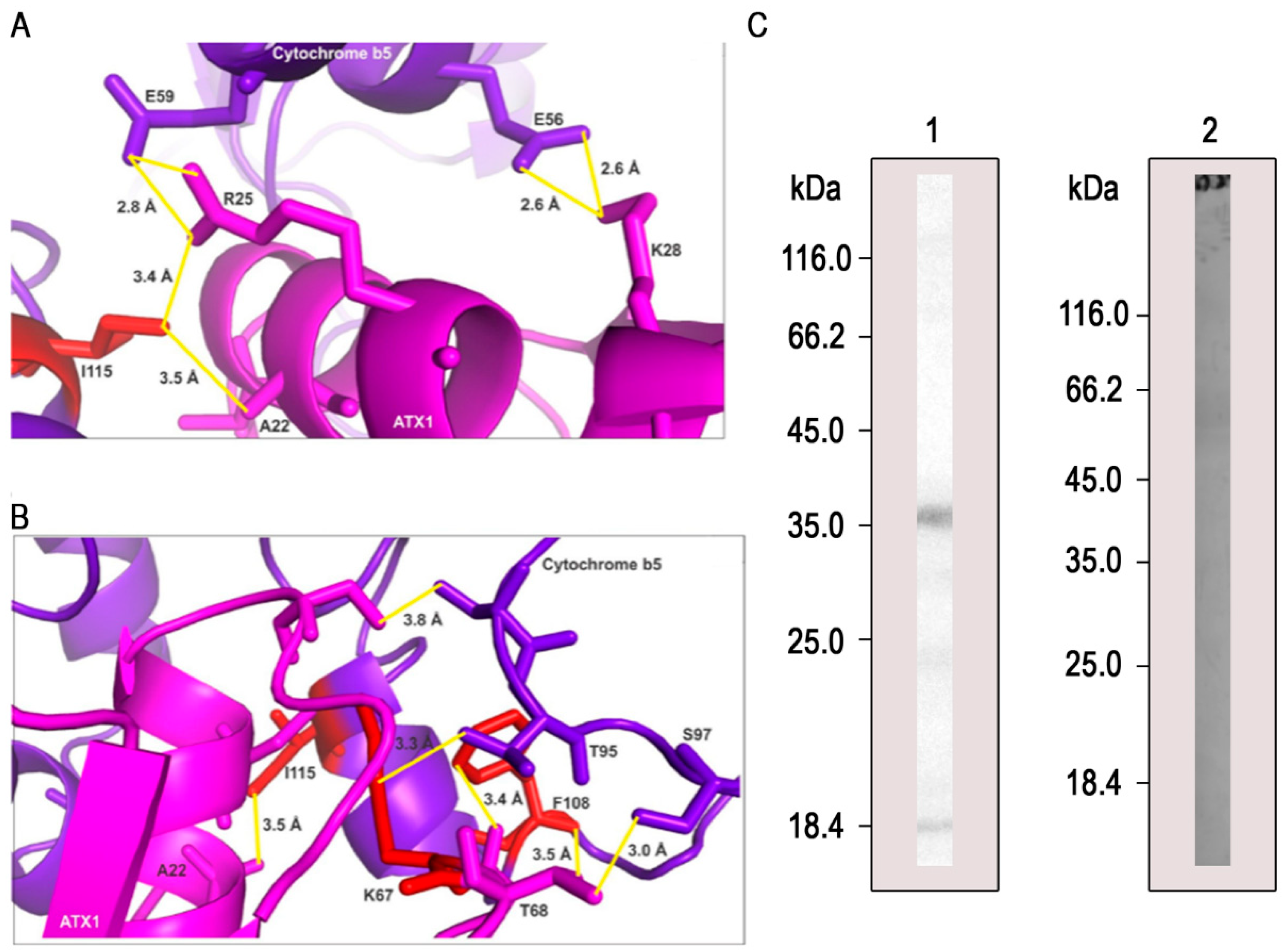
3.2. Pull-Down Validation by Western Blot Analysis
3.3. In Silico Seek for PbAtx1 Interaction Network
3.4. Molecular Modeling of PbAtx1 and PbCyb5 Resulted in Stable and Conserved Conformational Structures
3.5. PbAtx1 Interacts with PbCyb5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gonzalez, A.; Hernandez, O. New Insights into a Complex Fungal Pathogen: The Case of Paracoccidioides spp. Yeast 2015, 33, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caza, M.; Kronstad, J.W. Shared and Distinct Mechanisms of Iron Acquisition by Bacterial and Fungal Pathogens of Humans. Front. Cell. Infect. Microbiol. 2013, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.G.; Schrank, A.; Bailão, E.F.L.C.; Bailão, A.M.; Borges, C.L.; Staats, C.C.; Parente, J.A.; Pereira, M.; Salem-Izacc, S.M.; Mendes-Giannini, M.J.S.; et al. The Homeostasis of Iron, Copper, and Zinc in Paracoccidioides Brasiliensis, Cryptococcus Neoformans Var. Grubii, and Cryptococcus Gattii: A Comparative Analysis. Front. Microbiol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevitt, T.; Öhrvik, H.; Thiele, D.J. Charting the Travels of Copper in Eukaryotes from Yeast to Mammals. Biochim. Et Biophys. Acta 2013, 1823, 1580–1593. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for Copper Acquisition, Distribution and Regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Lin, S.J.; Culotta, V.C. The ATX1 Gene of Saccharomyces Cerevisiae Encodes a Small Metal Homeostasis Factor That Protects Cells against Reactive Oxygen Toxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-J.; Pufahl, R.A.; Dancis, A.; O’Halloran, T.V.; Culotta, V.C. A Role for the Saccharomyces Cerevisiae ATX1 Gene in Copper Trafficking and Iron Transport *. J. Biol. Chem. 1997, 272, 9215–9220. [Google Scholar] [CrossRef] [Green Version]
- Cankorur-Cetinkaya, A.; Eraslan, S.; Kirdar, B. Transcriptomic Response of Yeast Cells to ATX1 Deletion under Different Copper Levels. BMC Genom. 2016, 17, 489. [Google Scholar] [CrossRef] [Green Version]
- Walton, F.J.; Idnurm, A.; Heitman, J. Novel Gene Functions Required for Melanization of the Human Pathogen Cryptococcus Neoformans. Mol. Microbiol. 2005, 57, 1381–1396. [Google Scholar] [CrossRef]
- Anabosi, D.; Meir, Z.; Shadkchan, Y.; Handelman, M.; Abou-Kandil, A.; Yap, A.; Urlings, D.; Gold, M.S.; Krappmann, S.; Haas, H.; et al. Transcriptional Response of Aspergillus Fumigatus to Copper and the Role of the Cu Chaperones. Virulence 2021, 12, 2186–2200. [Google Scholar] [CrossRef]
- Shikanai-Yasuda, M.A.; Mendes, R.P.; Colombo, A.L.; de Queiroz-Telles, F.; Kono, A.S.G.; Paniago, A.M.M.; Nathan, A.; do Valle, A.C.F.; Bagagli, E.; Benard, G.; et al. Brazilian Guidelines for the Clinical Management of Paracoccidioidomycosis. Rev. Da Soc. Bras. De Med. Trop. 2017, 50, 715–740. [Google Scholar] [CrossRef] [PubMed]
- Giusiano, G. The Trojan Horse Model in Paracoccidioides: A Fantastic Pathway to Survive Infecting Human Cells. Front. Cell. Infect. Microbiol. 2021, 10, 605679. [Google Scholar] [CrossRef] [PubMed]
- Besold, A.N.; Culbertson, E.M.; Culotta, V.C. The Yin and Yang of Copper During Infection. J. Biol. Inorg. Chem. 2016, 21, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Santamarina, S.; Thiele, D.J. Copper at the Fungal Pathogen-Host Axis *. J. Biol. Chem. 2015, 290, 18945–18953. [Google Scholar] [CrossRef] [Green Version]
- Petito, G.; De Curcio, J.S.; Pereira, M.; Bailão, A.M.; Paccez, J.; Tristão, G.B.; de Morais, C.O.B.; De Souza, M.V.; Santos, A.J.D.C.M.; Fontes, W.; et al. Metabolic Adaptation of Paracoccidioides Brasiliensis in Response to in Vitro Copper Deprivation. Front. Microbiol. 2020, 11, 1834. [Google Scholar] [CrossRef]
- Portis, I.G.; de Sousa Lima, P.; Paes, R.A.; Oliveira, L.N.; Pereira, C.A.; Parente-Rocha, J.A.; Pereira, M.; Nosanchuk, J.D.; de Almeida Soares, C.M. Copper Overload in Paracoccidioides Lutzii Results in the Accumulation of Ergosterol and Melanin. Microbiol. Res. 2020, 239, 126524. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Ahmad, F.; Ahmad, N.; Flynn, D.C.; Kumar, R. Protein-Protein Interactions: Principles, Techniques, and Their Potential Role in New Drug Development. J. Biomol. Struct. Dyn. 2011, 28, 929–938. [Google Scholar] [CrossRef]
- Fava Netto, C. Contribuição Para o Estudo Imunológico Da Blastomicose de Lutz (Blastomicose Sul-Americana). Rev. Do Inst. Adolfo Lutz 1961, 21, 99–194. [Google Scholar]
- Murad, A.M.; Souza, G.H.M.F.; Garcia, J.S.; Rech, E.L. Detection and Expression Analysis of Recombinant Proteins in Plant-Derived Complex Mixtures Using NanoUPLC-MSE. J. Sep. Sci. 2011, 34, 2618–2630. [Google Scholar] [CrossRef] [Green Version]
- Geromanos, S.J.; Vissers, J.P.C.; Silva, J.C.; Dorschel, C.A.; Li, G.-Z.; Gorenstein, M.V.; Bateman, R.H.; Langridge, J.I. The Detection, Correlation, and Comparison of Peptide Precursor and Product Ions from Data Independent LC-MS with Data Dependant LC-MS/MS. Proteomics 2009, 9, 1683–1695. [Google Scholar] [CrossRef]
- de Souza, A.F.; Pigosso, L.L.; Silva, L.O.S.; Galo, I.D.C.; Paccez, J.D.; e Silva, K.S.F.; de Oliveira, M.A.P.; Pereira, M.; de Almeida Soares, C.M. Iron Deprivation Modulates the Exoproteome in Paracoccidioides Brasiliensis. Front. Cell. Infect. Microbiol. 2022, 12, 903070. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swendsen, R.H.; Wang, J.-S. Replica Monte Carlo Simulation of Spin-Glasses. Phys. Rev. Lett. 1986, 57, 2607–2609. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y. LOMETS: A Local Meta-Threading-Server for Protein Structure Prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. SPICKER: A Clustering Approach to Identify near-Native Protein Folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–Ligand Binding Site Recognition Using Complementary Binding-Specific Substructure Comparison and Sequence Profile Alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-Based Maximal Affinity Model Predicts Small-Molecule Druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Zhu, X.; Mitchell, J.C. KFC2: A Knowledge-Based Hot Spot Prediction Method Based on Interface Solvation, Atomic Density, and Plasticity Features. Proteins Struct. Funct. Bioinform. 2011, 79, 2671–2683. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Spinelli, R.; Scarano, V.; Cavallo, L.; Oliva, R. COCOMAPS: A Web Application to Analyze and Visualize Contacts at the Interface of Biomolecular Complexes. Bioinformatics 2011, 27, 2915–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, E.G.A.; Weber, S.S.; Báo, S.N.; Pereira, L.A.; Bailão, A.M.; Borges, C.L.; De Almeida Soares, C.M. Analysis of Paracoccidioides Secreted Proteins Reveals Fructose 1,6-Bisphosphate Aldolase as a Plasminogen-Binding Protein. BMC Microbiol. 2015, 15, 53. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.G.; de Curcio, J.S.; Silva-Bailão, M.G.; Lima, R.M.; Tomazett, M.V.; de Souza, A.F.; Cruz-Leite, V.R.M.; Sbaraini, N.; Bailão, A.M.; Rodrigues, F.; et al. Molecular Characterization of Siderophore Biosynthesis in Paracoccidioides Brasiliensis. IMA Fungus 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Tottey, S.; Rondet, S.A.M.; Borrelly, G.P.M.; Robinson, P.J.; Rich, P.R.; Robinson, N.J. A Copper Metallochaperone for Photosynthesis and Respiration Reveals Metal-Specific Targets, Interaction with an Importer, and Alternative Sites for Copper Acquisition. J. Biol. Chem. 2002, 277, 5490–5497. [Google Scholar] [CrossRef] [Green Version]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate Synthetase from the Urea Cycle to the Citrulline–NO Cycle. Eur. J. Biochem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef]
- Dietl, A.-M.; Binder, U.; Bauer, I.; Shadkchan, Y.; Osherov, N.; Haas, H. Arginine Auxotrophy Affects Siderophore Biosynthesis and Attenuates Virulence of Aspergillus Fumigatus. Genes 2020, 11, 423. [Google Scholar] [CrossRef] [Green Version]
- Festa, R.A.; Thiele, D.J. Copper: An Essential Metal in Biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef] [Green Version]
- Portnoy, M.E.; Rosenzweig, A.C.; Rae, T.; Huffman, D.L.; O’Halloran, T.V.; Culotta, V.C. Structure-Function Analyses of the ATX1 Metallochaperone*. J. Biol. Chem. 1999, 274, 15041–15045. [Google Scholar] [CrossRef] [Green Version]
- Karlenius, T.C.; Tonissen, K.F. Thioredoxin and Cancer: A Role for Thioredoxin in All States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Brose, J.; La Fontaine, S.; Wedd, A.G.; Xiao, Z. Redox Sulfur Chemistry of the Copper Chaperone Atox1 Is Regulated by the Enzyme Glutaredoxin 1, the Reduction Potential of the Glutathione Couple GSSG/2GSH and the Availability of Cu(i)†. Metallomics 2014, 6, 793–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abicht, H.K.; Schärer, M.A.; Quade, N.; Ledermann, R.; Mohorko, E.; Capitani, G.; Hennecke, H.; Glockshuber, R. How Periplasmic Thioredoxin TlpA Reduces Bacterial Copper Chaperone ScoI and Cytochrome Oxidase Subunit II (CoxB) Prior to Metallation *♦. J. Biol. Chem. 2014, 289, 32431–32444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ren, Y.; Gao, L.; Gu, H.; Lua, L. Electron Donor Cytochrome B5 Is Required for Hyphal Tip Accumulation of Sterol-Rich Plasma Membrane Domains and Membrane Fluidity in Aspergillus Fumigatus. Appl. Environ. Microbiol. 2021, 87, e02571-20. [Google Scholar] [CrossRef] [PubMed]
- Misslinger, M.; Gsaller, F.; Hortschansky, P.; Müller, C.; Bracher, F.; Bromley, M.J.; Haas, H. The Cytochrome B5 CybE Is Regulated by Iron Availability and Is Crucial for Azole Resistance in A. fumigatus. Metallomics 2017, 9, 1655–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebenmorgen, T.; Zacharias, M. Evaluation of Predicted Protein–Protein Complexes by Binding Free Energy Simulations. J. Chem. Theory Comput. 2019, 15, 2071–2086. [Google Scholar] [CrossRef]
- Bull, P.C.; Cox, D.W. Wilson Disease and Menkes Disease: New Handles on Heavy-Metal Transport. Trends Genet. 1994, 10, 246–252. [Google Scholar] [CrossRef]
- Kortemme, T.; Baker, D. A Simple Physical Model for Binding Energy Hot Spots in Protein–Protein Complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Zhang, X. Identification of Hot Regions in Hub Protein–Protein Interactions by Clustering and PPRA Optimization. BMC Med. Inform. Decis. Mak. 2021, 21, 143. [Google Scholar] [CrossRef]
- Pufahl, R.A.; Singer, C.P.; Peariso, K.L.; Lin, S.J.; Schmidt, P.J.; Fahrni, C.J.; Cizewski Culotta, V.; Penner-Hahn, J.E.; O’Halloran, T.V. Metal Ion Chaperone Function of the Soluble Cu(I) Receptor Atx1. Science 1997, 278, 853–856. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession a | Description | Score | Sequence Coverage | Peptides b |
|---|---|---|---|---|
| PADG_00888 | Argininosuccinate synthase | 797.87 | 14.22 | 6 |
| PADG_01886 | Adenosylhomocysteinase | 1375.59 | 27.52 | 14 |
| PADG_04167 | Aspartyl aminopeptidase | 863.77 | 21.96 | 13 |
| PADG_03449 | Isopentenyl-diphosphate delta-isomerase | 872.73 | 13.28 | 8 |
| PADG_00221 | Short-chain dehydrogenase | 1357.70 | 21.4 | 9 |
| PADG_05081 | Aldehyde dehydrogenase | 1362.79 | 30.8 | 16 |
| PADG_00206 | Mitochondrial zinc maintenance protein 1, mitochondrial | 2151.42 | 10.48 | 3 |
| PADG_01551 | Thioredoxin reductase | 1759.66 | 22.35 | 8 |
| PADG_03161 | Thioredoxin | 906.63 | 33.73 | 13 |
| PADG_02181 | HAD-superfamily hydrolase | 718.42 | 24.36 | 9 |
| PADG_00128 | Tubulin alpha-2 chain | 655.55 | 21.29 | 9 |
| PADG_07422 | Serine proteinase | 1204.78 | 5.66 | 4 |
| PADG_00688 | F-type H+-transporting ATPase subunit H | 1610.02 | 36.36 | 5 |
| PADG_03559 | Cytochrome-b5 | 955.84 | 35.51 | 3 |
| PADG_07883 | hypothetical protein | 790.64 | 35.8 | 4 |
| PADG_04855 | hypothetical protein | 810.68 | 20.31 | 10 |
| PADG_05110 | hypothetical protein | 703.68 | 19.5 | 6 |
| PADG_00449 | hypothetical protein | 1232.59 | 13.7 | 16 |
| PADG_03788 | hypothetical protein | 811.28 | 15.03 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Carvalho Júnior, M.A.B.; Silva, L.O.S.; Pigosso, L.L.; de Souza, A.F.; Lugo, D.E.M.; Moraes, D.; Freitas e Silva, K.S.; Pereira, M.; Soares, C.M.d.A. Molecular Interactions of the Copper Chaperone Atx1 of Paracoccidioides brasiliensis with Fungal Proteins Suggest a Crosstalk between Iron and Copper Homeostasis. Microorganisms 2023, 11, 248. https://doi.org/10.3390/microorganisms11020248
de Carvalho Júnior MAB, Silva LOS, Pigosso LL, de Souza AF, Lugo DEM, Moraes D, Freitas e Silva KS, Pereira M, Soares CMdA. Molecular Interactions of the Copper Chaperone Atx1 of Paracoccidioides brasiliensis with Fungal Proteins Suggest a Crosstalk between Iron and Copper Homeostasis. Microorganisms. 2023; 11(2):248. https://doi.org/10.3390/microorganisms11020248
Chicago/Turabian Stylede Carvalho Júnior, Marcos Antonio Batista, Lana O’Hara Souza Silva, Laurine Lacerda Pigosso, Aparecido Ferreira de Souza, Danize Eukales Menezes Lugo, Dayane Moraes, Kleber Santiago Freitas e Silva, Maristela Pereira, and Célia Maria de Almeida Soares. 2023. "Molecular Interactions of the Copper Chaperone Atx1 of Paracoccidioides brasiliensis with Fungal Proteins Suggest a Crosstalk between Iron and Copper Homeostasis" Microorganisms 11, no. 2: 248. https://doi.org/10.3390/microorganisms11020248