
Introduction, Dispersal, and Predominance of SARS-CoV-2 Delta Variant in Rio Grande do Sul, Brazil: A Retrospective Analysis
 , , , , ,
, , , , ,  and add
Show full author list
and add
Show full author list
Abstract
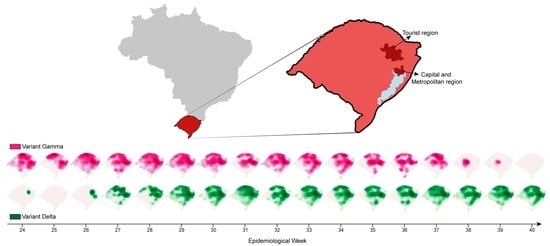
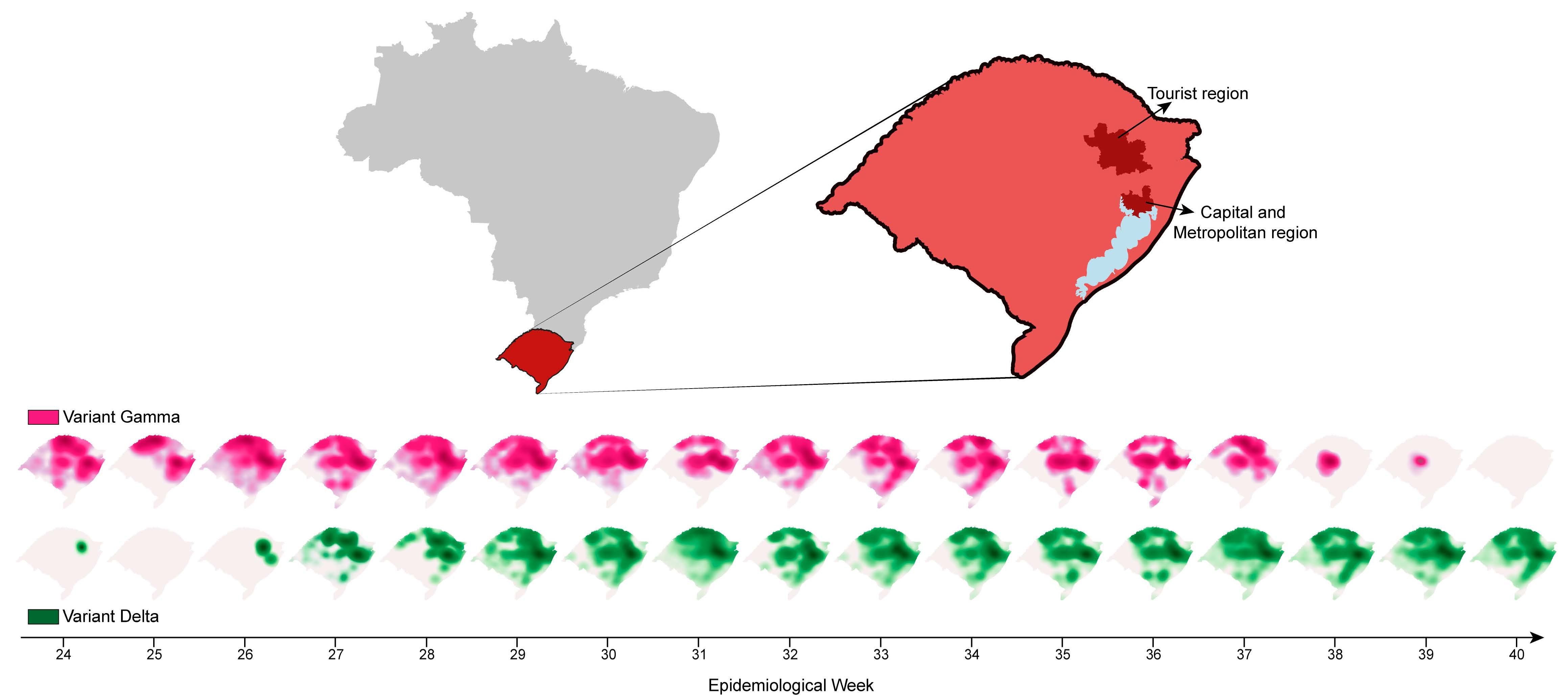
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Epidemiological and Clinical Data
2.3. Sample Screening and Genome Sequencing
2.3.1. Viral Genome Assembly
2.3.2. Lineage Identification
2.3.3. Single Nucleotide Polymorphism Identification
2.4. Spatio-Temporal Analysis
2.5. Phylogenetic Analysis
2.5.1. Data Sets
2.5.2. Analysis of Temporal Signal and Identification of Brazilian Clades
3. Results
3.1. COVID-19 Overview
3.2. Epidemiological Aspects of Delta Introduction Period
3.3. SARS-CoV-2 Delta Introduction and Dispersal
3.4. Single Nucleotide Polymorphism
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- CoVariants. Overview of Variants in Countries. Available online: https://covariants.org/per-country?variant=21A+%28Delta%29&variant=21I+%28Delta%29&variant=21J+%28Delta%29 (accessed on 1 November 2023).
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/publications/m/item/historical-working-definitions-and-primary-actions-for-sars-cov-2-variants (accessed on 1 November 2023).
- Volz, E.M.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Dos Santos, M.C.; Júnior, E.C.; Ferreira, J.A.; Barbagelata, L.S.; Da Silva, S.P.; Silva, A.M.; Cardoso, J.; Bedran, R.L.S.; Júnior, W.D.C.; Bezerra, D.A.M.; et al. First Reported Cases of SARS-CoV-2 Sub-Lineage B.1.617.2 in Brazil: An Outbreak in a Ship and Alert for Spread. Available online: https://virological.org/t/first-reported-cases-of-sars-cov-2-sub-lineage-b-1-617-2-in-brazil-an-outbreak-in-a-ship-and-alert-for-spread/706 (accessed on 3 October 2022).
- Lamarca, A.P.; de Almeida, L.G.P.; da Silva Francisco, R.; Cavalcante, L.; Machado, D.T.; Brustolini, O.; Gerber, A.L.; de C Guimarães, A.P.; Policarpo, C.; da Silva de Oliveira, G.; et al. Genomic Surveillance Tracks the First Community Outbreak of the SARS-CoV-2 Delta (B.1.617.2) Variant in Brazil. J. Virol. 2022, 96, e01228-21. [Google Scholar] [CrossRef]
- Arantes, I.; Naveca, F.G.; Gräf, T.; Miyajima, F.; Faoro, H.; Wallau, G.L.; Delatorre, E.; Appolinario, L.R.; Pereira, E.C.; Venas, T.M.M.; et al. Emergence and Spread of the SARS-CoV-2 Variant of Concern Delta across Different Brazilian Regions. Microbiol. Spectr. 2022, 10, e02641-21. [Google Scholar] [CrossRef]
- Rio Grande do Sul State Government. Geography of Rio Grande Do Sul State. Available online: https://www.estado.rs.gov.br/geografia (accessed on 10 August 2022).
- Instituto Brasileiro de Geografia e Estatística (IBGE). Border Strip Municipalities and Twin Cities. Available online: https://www.ibge.gov.br/geociencias/organizacao-do-territorio/estrutura-territorial/24073-municipios-da-faixa-de-fronteira.html?=&t=acesso-ao-produto (accessed on 10 August 2022).
- Instituto Brasileiro de Geografia e Estatística (IBGE). Demographic Census 2010. Available online: https://cidades.ibge.gov.br/brasil/rs/pesquisa/23/25207?tipo=ranking (accessed on 10 August 2022).
- Parana State Govenment. Com 6412%, Paraná Alcança Maior Participação Da História No PIB Nacional. Available online: https://www.aen.pr.gov.br/Noticia/Com-6412-Parana-alcanca-maior-participacao-da-historia-no-PIB-nacional (accessed on 26 September 2023).
- Rio Grande do Sul State Government. Características Gerais—Atlas Socioeconômico Do Rio Grande Do Sul. Available online: https://atlassocioeconomico.rs.gov.br/caracteristicas-gerais (accessed on 26 September 2023).
- State Health Department of Rio Grande do Sul. Coronavirus Dashboard. Available online: https://ti.saude.rs.gov.br/covid19/ (accessed on 26 September 2023).
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Subedi, S.; Olson, R.; Davis, J.J.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; Reppond, K.; et al. Sequence Analysis of 20,453 Severe Acute Respiratory Syndrome Coronavirus 2 Genomes from the Houston Metropolitan Area Identifies the Emergence and Widespread Distribution of Multiple Isolates of All Major Variants of Concern. Am. J. Pathol. 2021, 191, 983–992. [Google Scholar] [CrossRef]
- Dhawan, M.; Sharma, A.; Priyanka; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta Variant (B.1.617.2) of SARS-CoV-2: Mutations, Impact, Challenges and Possible Solutions. Hum. Vaccin. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef]
- Ferreira, I.A.T.M.; Kemp, S.A.; Datir, R.; Saito, A.; Meng, B.; Rakshit, P.; Takaori-Kondo, A.; Kosugi, Y.; Uriu, K.; Kimura, I.; et al. SARS-CoV-2 B.1.617 Mutations L452R and E484Q Are Not Synergistic for Antibody Evasion. J. Infect. Dis. 2021, 224, 989–994. [Google Scholar] [CrossRef]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Sidarovich, A.; Winkler, M.S.; Schulz, S.; Jäck, H.M.; Stankov, M.V.; Behrens, G.M.N.; et al. Delta Variant (B.1.617.2) Sublineages Do Not Show Increased Neutralization Resistance. Cell. Mol. Immunol. 2021, 18, 2557–2559. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.W.; Jamrozy, D. Genomics and Epidemiological Surveillance. Nat. Rev. Microbiol. 2020, 18, 478. [Google Scholar] [CrossRef]
- Hu, T.; Li, J.; Zhou, H.; Li, C.; Holmes, E.C.; Shi, W. Bioinformatics Resources for SARS-CoV-2 Discovery and Surveillance. Brief. Bioinform. 2021, 22, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global Landscape of SARS-CoV-2 Genomic Surveillance and Data Sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]
- State Health Department of Rio Grande do Sul. COVID-19 Immunization Monitoring. Available online: https://vacina.saude.rs.gov.br/ (accessed on 5 October 2022).
- Artic Network. nCov-2019 Sequencing Protocol v3 (LoCost). Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bp2l6n26rgqe/v3?version_warning=no (accessed on 18 August 2022).
- Artic Network. nCoV-2019 Novel Coronavirus Bioinformatics Protocol. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 18 August 2022).
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab00. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Dezordi, F.Z.; da Silva Neto, A.M.; de Lima Campos, T.; Jeronimo, P.M.C.; Aksenen, C.F.; Almeida, S.P.; Wallau, G.L. ViralFlow: A Versatile Automated Workflow for SARS-CoV-2 Genome Assembly, Lineage Assignment, Mutations and Intrahost Variant Detection. Viruses 2022, 14, 217. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- GISAID EpiCoV Database. Available online: https://gisaid.org/ (accessed on 20 October 2022).
- Council of Municipal Health Departments of Rio Grande do Sul. Health Regions. Available online: https://www.cosemsrs.org.br/regioes-de-saude (accessed on 3 October 2022).
- QGIS Development Team. QGIS: A Free and Open Source Geographic Information System. Available online: https://qgis.org/en/site/ (accessed on 3 October 2022).
- Instituto Brasileiro de Geografia e Estatística (IBGE). Rio Grande Do Sul Cartographic Base. Available online: https://geoftp.ibge.gov.br/cartas_e_mapas/bases_cartograficas_continuas/bc100/rio_grande_do_sul/ (accessed on 3 October 2022).
- Fonseca, P.L.C.; Moreira, F.R.R.; De Souza, R.M.; Guimarães, N.R.; Carvalho, N.O.; Adelino, T.E.R.; Alves, H.J.; Alvim, L.B.; Candido, D.S.; Coelho, H.P.; et al. Tracking the Turnover of SARS-CoV-2 VOCs Gamma to Delta in a Brazilian State (Minas Gerais) with a High-Vaccination Status. Virus Evol. 2022, 8, veac064. [Google Scholar] [CrossRef] [PubMed]
- Brazilian Government. Coronavirus Dashboard. Available online: https://covid.saude.gov.br/ (accessed on 20 October 2022).
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Some Mathematical Questions in Biology: DNA Sequence Analysis. Stat. Med. 1989, 8, 523–524. [Google Scholar] [CrossRef]
- Yang, Z. Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 20 October 2022).
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Gularte, J.S.; da Silva, M.S.; Mosena, A.C.S.; Demoliner, M.; Hansen, A.W.; Filippi, M.; de Abreu Góes Pereira , V.M.; Heldt, F.H.; Weber, M.N.; de Almeida, P.R.; et al. Early Introduction, Dispersal and Evolution of Delta SARS-CoV-2 in Southern Brazil, Late Predominance of AY.99.2 and AY.101 Related Lineages. Virus Res. 2022, 311, 198702. [Google Scholar] [CrossRef]
- Presidency of the Federative Republic of Brazil. Law Number 13,979, of 6 February 2020. Available online: http://www.planalto.gov.br/ccivil_03/_ato2019-2022/2020/lei/L13979.htm (accessed on 3 October 2022).
- Elliott, P.; Haw, D.; Wang, H.; Eales, O.; Walters, C.E.; Ainslie, K.E.C.; Atchison, C.; Fronterre, C.; Diggle, P.J.; Page, A.J.; et al. Exponential Growth, High Prevalence of SARS-CoV-2, and Vaccine Effectiveness Associated with the Delta Variant. Science 2021, 374, eabl9551. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Salman, M.; Haider, S.A.; Badar, N.; Rehman, Z.; Ammar, M.; Rana, M.S.; Ali, Q. Genomic Surveillance Reveals the Detection of SARS-CoV-2 Delta, Beta, and Gamma VOCs during the Third Wave in Pakistan. J. Med. Virol. 2022, 94, 1115–1129. [Google Scholar] [CrossRef]
- Yakovleva, A.; Kovalenko, G.; Redlinger, M.; Liulchuk, M.G.; Bortz, E.; Zadorozhna, V.I.; Scherbinska, A.M.; Wertheim, J.O.; Goodfellow, I.; Meredith, L.; et al. Tracking SARS-COV-2 Variants Using Nanopore Sequencing in Ukraine in 2021. Sci. Rep. 2022, 12, 15749. [Google Scholar] [CrossRef]
- de Menezes Mayer, A.; Gröhs Ferrareze, P.A.; de Oliveira, L.F.V.; Gregianini, T.S.; Neves, C.L.A.M.; Caldana, G.D.; Kmetzsch, L.; Thompson, C.E. Genomic Characterization and Molecular Evolution of SARS-CoV-2 in Rio Grande Do Sul State, Brazil. Virology 2023, 582, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ferrareze, P.A.G.; Cybis, G.B.; de Oliveira, L.F.V.; Zimerman, R.A.; Schiavon, D.E.B.; Peter, C.; Thompson, C.E. Intense P.1 (Gamma) Diversification Followed by Rapid Delta Substitution in Southern Brazil: A SARS-CoV-2 Genomic Epidemiology Study. Microbes Infect. 2023, 10, 105216. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.R.R.; D’Arc, M.; Mariani, D.; Herlinger, A.L.; Schiffler, F.B.; Rossi, Á.D.; Leitão, I.D.C.; Miranda, T.D.S.; Cosentino, M.A.C.; Tôrres, M.C.D.P.; et al. Epidemiological Dynamics of SARS-CoV-2 VOC Gamma in Rio de Janeiro, Brazil. Virus Evol. 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- de Souza, U.J.B.; Dos Santos, R.N.; de Melo, F.L.; Belmok, A.; Galvão, J.D.; de Rezende, T.C.V.; Cardoso, F.D.P.; Carvalho, R.F.; da Silva Oliveira, M.; Junior, J.C.R.; et al. Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil. Viruses 2022, 14, 659. [Google Scholar] [CrossRef] [PubMed]
- Sgorlon, G.; da Silva Queiroz, J.A.; Roca, T.P.; da Silva, A.M.P.; Gasparelo, N.W.F.; Teixeira, K.S.; da Nóbrega Oliveira, A.S.; de Melo Mendonça, A.L.F.; Maia, A.C.S.; Pereira, S.D.S.; et al. Clinical and Epidemiological Aspects of Delta and Gamma SARS-CoV-2 Variant of Concern from the Western Brazilian Amazon. Mem. Inst. Oswaldo Cruz 2022, 117, 117. [Google Scholar] [CrossRef] [PubMed]
- Pinho, C.T.; Vidal, A.F.; Costa Negri Rocha, T.; Oliveira, R.R.M.; Clara da Costa Barros, M.; Closset, L.; Azevedo-Pinheiro, J.; Braga-da-Silva, C.; Santos Silva, C.; Magalhães, L.L.; et al. Transmission Dynamics of SARS-CoV-2 Variants in the Brazilian State of Pará. Front. Public Health 2023, 11, 11. [Google Scholar] [CrossRef]
- Silva, J.P.; de Lima, A.B.; Alvim, L.B.; Malta, F.S.V.; Mendonça, C.P.T.B.; Fonseca, P.L.C.; Moreira, F.R.R.; Queiroz, D.C.; Ferreira, J.G.G.; Ferreira, A.C.S.; et al. Delta Variant of SARS-CoV-2 Replacement in Brazil: A National Epidemiologic Surveillance Program. Viruses 2022, 14, 847. [Google Scholar] [CrossRef]
- Giovanetti, M.; Fonseca, V.; Wilkinson, E.; Tegally, H.; San, E.J.; Althaus, C.L.; Xavier, J.; Slavov, S.N.; Viala, V.L.; Lima, A.R.J.; et al. Replacement of the Gamma by the Delta Variant in Brazil: Impact of Lineage Displacement on the Ongoing Pandemic. Virus Evol. 2022, 8, veac024. [Google Scholar] [CrossRef]
- Patané, J.; Viala, V.; Lima, L.; Martins, A.; Barros, C.; Bernardino, J.; Moretti, D.; Slavov, S.; Santos, R.; Rodrigues, E.; et al. SARS-CoV-2 Delta Variant of Concern in Brazil—Multiple Introductions, Communitary Transmission, and Early Signs of Local Evolution. medRxiv 2021. [Google Scholar] [CrossRef]
- Saifi, S.; Ravi, V.; Sharma, S.; Swaminathan, A.; Chauhan, N.S.; Pandey, R. SARS-CoV-2 VOCs, Mutational Diversity and Clinical Outcome: Are They Modulating Drug Efficacy by Altered Binding Strength? Genomics 2022, 114, 110466. [Google Scholar] [CrossRef]
- Shen, L.; Triche, T.J.; Dien Bard, J.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD Mutation G142D in SARS-CoV-2 Delta VOC Lineages Is Associated with Frequent Back Mutations, Increased Viral Loads, and Immune Evasion. medRxiv 2021. [Google Scholar] [CrossRef]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and Genomics of SARS-CoV-2: A Review of the Literature with the Special Focus on Genetic Diversity and SARS-CoV-2 Genome Detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; Tenoever, B.R.; Sanjana, N.E. The Spike D614G Mutation Increases SARS-CoV-2 Infection of Multiple Human Cell Types. Elife 2021, 10, e65365. [Google Scholar] [CrossRef]
- Groves, D.C.; Rowland-Jones, S.L.; Angyal, A. The D614G Mutations in the SARS-CoV-2 Spike Protein: Implications for Viral Infectivity, Disease Severity and Vaccine Design. Biochem. Biophys. Res. Commun. 2021, 538, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G Spike Mutation Increases Entry Efficiency with Enhanced ACE2-Binding Affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef]
- Ogawa, J.; Zhu, W.; Tonnu, N.; Singer, O.; Hunter, T.; Ryan, A.L.; Pao, G.M. The D614G Mutation in the SARS-CoV2 Spike Protein Increases Infectivity in an ACE2 Receptor Dependent Manner. bioRxiv 2020, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Winger, A.; Caspari, T. The Spike of Concern-The Novel Variants of SARS-CoV-2. Viruses 2021, 13, 1002. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Lamarca, A.P.; de Almeida, L.G.P.P.; da Silva Francisco, R.; Cavalcante, L.; Brustolini, O.; Gerber, A.L.; de C Guimarães, A.P.; de Oliveira, T.H.; Nascimento, É.R.D.S.; Policarpo, C.; et al. Phylodynamic Analysis of SARS-CoV-2 Spread in Rio de Janeiro, Brazil, Highlights How Metropolitan Areas Act as Dispersal Hubs for New Variants. Microb. Genom. 2022, 8, 000859. [Google Scholar] [CrossRef]
- te Velthuis, A.J.W.; Arnold, J.J.; Cameron, C.E.; van den Worm, S.H.E.; Snijder, E.J. The RNA Polymerase Activity of SARS-Coronavirus Nsp12 Is Primer Dependent. Nucleic Acids Res. 2010, 38, 203–214. [Google Scholar] [CrossRef]
- Zhang, B.Z.; Hu, Y.; Chen, L.; Yau, T.; Tong, Y.; Hu, J.; Cai, J.; Chan, K.H.; Dou, Y.; Deng, J.; et al. Mining of Epitopes on Spike Protein of SARS-CoV-2 from COVID-19 Patients. Cell Res. 2020, 30, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Mishra, T.; Dalavi, R.; Joshi, G.; Kumar, A.; Pandey, P.; Shukla, S.; Mishra, R.K.; Chande, A. SARS-CoV-2 Spike E156G/Δ157-158 Mutations Contribute to Increased Infectivity and Immune Escape. Life Sci. Alliance 2022, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody Evasion by the P.1 Strain of SARS-CoV-2. Cell 2021, 184, 2939–2954. [Google Scholar] [CrossRef]
- Padilha, D.A.; Filho, V.B.; Moreira, R.S.; Soratto, T.A.T.; Maia, G.A.; Christoff, A.P.; Barazzetti, F.H.; Schörner, M.A.; Ferrari, F.L.; Martins, C.L.; et al. Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil. Viruses 2022, 14, 695. [Google Scholar] [CrossRef]
- Fratev, F. N501Y and K417N Mutations in the Spike Protein of SARS-CoV-2 Alter the Interactions with Both HACE2 and Human-Derived Antibody: A Free Energy of Perturbation Retrospective Study. J. Chem. Inf. Model. 2021, 61, 6079–6084. [Google Scholar] [CrossRef]
- Mohammadi, M.; Shayestehpour, M.; Mirzaei, H. The Impact of Spike Mutated Variants of SARS-CoV2 [Alpha, Beta, Gamma, Delta, and Lambda] on the Efficacy of Subunit Recombinant Vaccines. Braz. J. Infect. Dis. 2021, 25, 1–9. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef] [PubMed]
- Adam, D. What Scientists Know about New, Fast-Spreading Coronavirus Variants. Nature 2021, 594, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 Spike L452R Variant Evades Cellular Immunity and Increases Infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Serdan, T.D.A.; Gorjao, R.; Masi, L.N.; Pithon-Curi, T.C.; Covas, D.T.; Curi, R.; Durigon, E.L. SARS-COV-2 Variants: Differences and Potential of Immune Evasion. Front. Cell. Infect. Microbiol. 2021, 11, 1401. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher Infectivity of the SARS-CoV-2 New Variants Is Associated with K417N/T, E484K, and N501Y Mutants: An Insight from Structural Data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361. [Google Scholar] [CrossRef]
- Golubchik, T.; Lythgoe, K.A.; Hall, M.; Ferretti, L.; Fryer, H.R.; Maclntyre-Cockett, G.; de Cesare, M.; Trebes, A.; Piazza, P.; Buck, D.; et al. Early Analysis of a Potential Link between Viral Load and the N501Y Mutation in the SARS-COV-2 Spike Protein. medRxiv 2021. [Google Scholar] [CrossRef]
- Islam, S.R.; Prusty, D.; Manna, S.K. Structural Basis of Fitness of Emerging SARS-COV-2 Variants and Considerations for Screening, Testing and Surveillance Strategy to Contain Their Threat. medRxiv 2021. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 Variants of Concern Link to Increased Spike Cleavage and Virus Transmission. Cell Host Microbe 2022, 30, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.A.; Medina, P.M. Temporal Changes in the Accessory Protein Mutations of SARS-CoV-2 Variants and Their Predicted Structural and Functional Effects. J. Med. Virol. 2022, 94, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid Mutations in SARS-CoV-2 Augment Replication and Pathogenesis. PLoS Pathog. 2022, 18, e1010627. [Google Scholar] [CrossRef]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid Mutations R203K/G204R Increase the Infectivity, Fitness, and Virulence of SARS-CoV-2. Cell Host Microbe 2021, 29, 1788–1801. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gamma Positive (n = 422) | Delta Positive (n = 648) | Others (n = 7) | |
|---|---|---|---|
| Age (median) | 43 | 41 | 37 |
| <18 (number) | 17 | 48 | 0 |
| 18–60 | 324 | 432 | 5 |
| >60 | 81 | 167 | 2 |
| Female (%) | 237 (56%) | 340 (52%) | 5 (71%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
y Castro, T.R.; Piccoli, B.C.; Vieira, A.A.; Casarin, B.C.; Tessele, L.F.; Salvato, R.S.; Gregianini, T.S.; Martins, L.G.; Resende, P.C.; Pereira, E.C.; et al. Introduction, Dispersal, and Predominance of SARS-CoV-2 Delta Variant in Rio Grande do Sul, Brazil: A Retrospective Analysis. Microorganisms 2023, 11, 2938. https://doi.org/10.3390/microorganisms11122938
y Castro TR, Piccoli BC, Vieira AA, Casarin BC, Tessele LF, Salvato RS, Gregianini TS, Martins LG, Resende PC, Pereira EC, et al. Introduction, Dispersal, and Predominance of SARS-CoV-2 Delta Variant in Rio Grande do Sul, Brazil: A Retrospective Analysis. Microorganisms. 2023; 11(12):2938. https://doi.org/10.3390/microorganisms11122938
Chicago/Turabian Styley Castro, Thaís Regina, Bruna C. Piccoli, Andressa A. Vieira, Bruna C. Casarin, Luíza F. Tessele, Richard S. Salvato, Tatiana S. Gregianini, Leticia G. Martins, Paola Cristina Resende, Elisa C. Pereira, and et al. 2023. "Introduction, Dispersal, and Predominance of SARS-CoV-2 Delta Variant in Rio Grande do Sul, Brazil: A Retrospective Analysis" Microorganisms 11, no. 12: 2938. https://doi.org/10.3390/microorganisms11122938