
Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral DNA Isolation
2.3. Detection of Viral DNA
2.4. Polymerase Gene Amplification
2.5. HBV Genotyping and Phylogenetic Analysis of HBV Isolates
2.6. Mutation Analysis
2.7. Statistical Analysis
3. Results
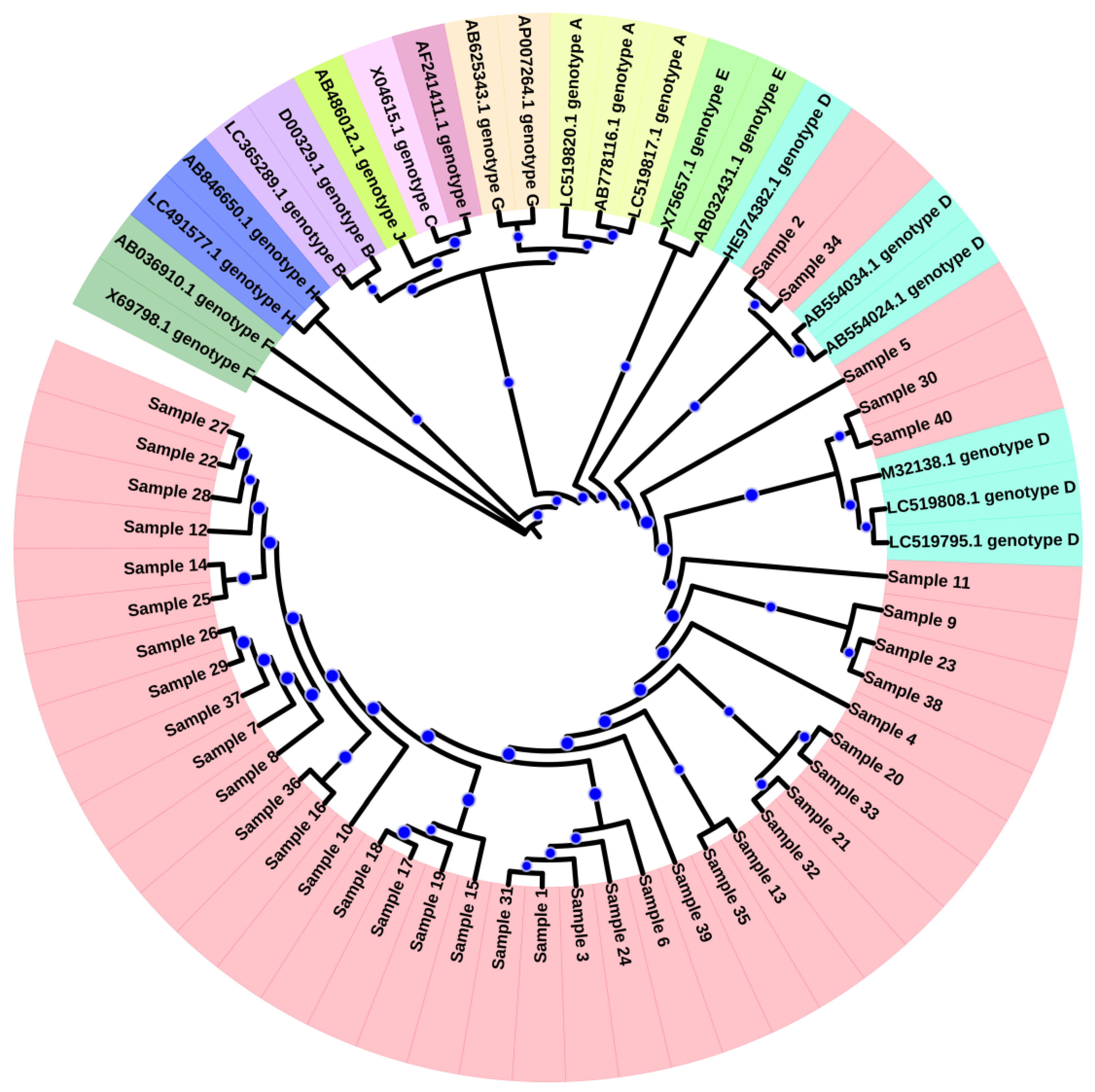
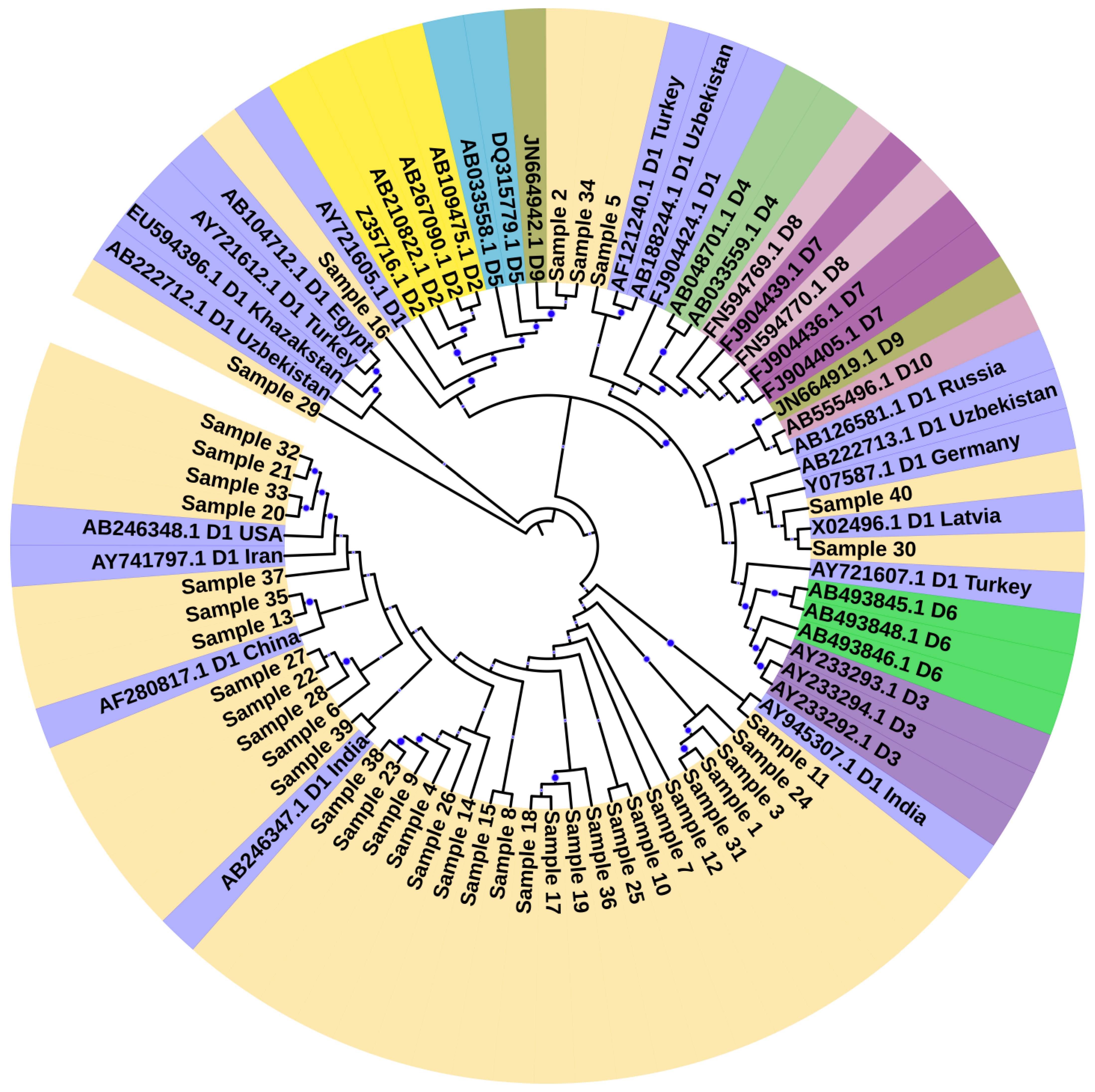
3.1. Genotyping and Phylogenetic Analysis
3.2. Drug Resistance Mutation
3.3. Genotypic Variants
3.4. Other Polymerase Gene Mutation
3.5. Small Surface Gene Mutation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dos Santos, M.I.M.A.; Pacheco, S.R.; Stocker, A.; Schinoni, M.I.; Paraná, R.; Reis, M.G.; Silva, L.K. Mutations associated with drug resistance and prevalence of vaccine escape mutations in patients with chronic hepatitis B infection. J. Med. Virol. 2017, 89, 1811–1816. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Tsai, K.N.; Ou, J.H.J. Pathogenicity and virulence of Hepatitis B virus. Virulence 2022, 13, 258–296. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadeq, D.W.; Taleb, S.A.; Zaied, R.E.; Fahad, S.M.; Smatti, M.K.; Rizeq, B.R.; Al Thani, A.A.; Yassine, H.M.; Nasrallah, G.K. Hepatitis B virus molecular epidemiology, host-virus interaction, coinfection, and laboratory diagnosis in the MENA region: An update. Pathogens 2019, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Hu, J. Hepatitis B virus virology and replication. In Hepatitis B Virus in Human Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–34. [Google Scholar]
- Pollack, J.R.; Ganem, D. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 1994, 68, 5579–5587. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Khan, S.A.; Hamayun, M.; Ali, M.; Idrees, M. Sequence variability of HCV 3a isolates based on core gene in patients from Lahore, Pakistan. Future Virol. 2019, 14, 641–653. [Google Scholar] [CrossRef]
- Ali, M.; Idrees, M.; Ali, L.; Hussain, A.; Rehman, I.U.; Saleem, S.; Afzal, S.; Butt, S. Hepatitis B virus in Pakistan: A systematic review of prevalence, risk factors, awareness status and genotypes. Virol. J. 2011, 8, 102. [Google Scholar] [CrossRef]
- Qureshi, H.; Bile, K.M.; Jooma, R.; Alam, S.E.; Afrid, H.U.R. Prevalence of hepatitis B and C viral infections in Pakistan: Findings of a national survey appealing for effective prevention and control measures. EMHJ-East. Mediterr. Health J. 2010, 16, 15–23. [Google Scholar] [CrossRef]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally occurring hepatitis B virus reverse transcriptase mutations related to potential antiviral drug resistance and liver disease progression. World J. Gastroenterol. 2018, 24, 1708. [Google Scholar] [CrossRef]
- Bartholomeusz, A.; Locarnini, S. Hepatitis B virus mutations associated with antiviral therapy. J. Med. Virol. 2006, 78, S52–S55. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.-M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Zhai, X.; Thai, H.; Campo, D.S.; Xia, G.; Ganova-Raeva, L.M.; Drobeniuc, J.; Khudyakov, Y.E. Evaluation of intra-host variants of the entire hepatitis B virus genome. PLoS ONE 2011, 6, e25232. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Ali, L.; Idrees, M.; Ali, M.; Rehman, I.U.; Hussain, A.; Afzal, S.; Butt, S.; Saleem, S.; Munir, S.; Badar, S. An overview of treatment response rates to various anti-viral drugs in Pakistani Hepatitis B Virus infected patients. Virol. J. 2011, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Zaidi, S.Z.; Shaukat, S.; Sharif, S.; Angez, M.; Naeem, A.; Saleha, S.; Butt, J.A.; Malik, S.A. Common Genotypes of Hepatitis B virus prevalent in Injecting drug abusers (addicts) of North West Frontier Province of Pakistan. Virol. J. 2007, 4, 63. [Google Scholar] [CrossRef]
- Mahmood, M.; Anwar, M.A.; Khanum, A.; Zaman, N.; Raza, A. Distribution and clinical significance of hepatitis B virus genotypes in Pakistan. BMC Gastroenterol. 2016, 16, 104. [Google Scholar] [CrossRef]
- Chien, R.N.; Liaw, Y.F. Current trend in antiviral therapy for chronic hepatitis B. Viruses 2022, 14, 434. [Google Scholar] [CrossRef]
- Liaw, Y.-F.; Chien, R.-N.; Yeh, C.-T.; Tsai, S.-L.; Chu, C.-M. Acute exacerbation and hepatitis B virus clearance after emergence of YMDD motif mutation during lamivudine therapy. Hepatology 1999, 30, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Girones, R.; Miller, R.H. Mutation rate of the hepadnavirus genome. Virology 1989, 170, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, Y.K.; Park, E.S.; Kim, K.H. Molecular diagnosis and treatment of drug-resistant hepatitis B virus. World J. Gastroenterol. 2014, 20, 5708–5720. [Google Scholar] [CrossRef] [PubMed]
- Doğan, M.; Müderrisoğlu, C.; Fincancı, M.; Ceylan, B.; Özdemir, G.E.; Polat, H. kronik hepatit b’de lamivudin direnci ve lamivudin direnci gelişimi üzerine etkili faktörler. Evaluation 2007, 39, 44. [Google Scholar]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Geipel, A.; Glebe, D.; Will, H.; Gerlich, W.H. Hepatitis B virus rtI233V mutation and resistance to adefovir. N. Engl. J. Med. 2014, 370, 1667–1668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Wei, M.; Zhang, C.; Xu, T.; Liu, L.; Xu, Z. Potential resistant mutations within HBV reverse transcriptase sequences in nucleos (t) ide analogues-experienced patients with hepatitis B virus infection. Sci. Rep. 2019, 9, 8078. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.; Anwar, M.A. Analysis of resistant mutations in reverse transcriptase domain of hepatitis B virus from patients from Islamabad, Pakistan. J. Unexplor. Med. Data 2017, 2, 60–64. [Google Scholar] [CrossRef]
- Marrone, A.; Zampino, R.; Karayannis, P.; Cirillo, G.; Cesaro, G.; Guerrera, B.; Ricciotti, R.; del Giudice, E.M.; Utili, R.; Adinolfi, L.E.; et al. Clinical reactivation during lamivudine treatment correlates with mutations in the precore/core promoter and polymerase regions of hepatitis B virus in patients with anti-hepatitis B e-positive chronic hepatitis. Aliment. Pharmacol. Ther. 2005, 22, 707–714. [Google Scholar] [CrossRef]
- Qin, B.; Pei, R.; He, T.; Huang, Z.; Pan, G.; Tu, C.; Lu, M.; Chen, X. Polymerase mutations rtN238R, rtT240Y and rtN248H of hepatitis B virus decrease susceptibility to adefovir. Chin. Sci. Bull. 2013, 58, 1760–1766. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Herbers, U.; Sheldon, J.; Luedde, T.; Trautwein, C.; Tacke, F. The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen–positive and hepatitis B e antigen–negative hepatitis B virus strains. Hepatology 2009, 49, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Wang, J.H.; Lu, S.N.; Hu, T.H.; Hung, C.H.; Chang, M.H.; Changchien, C.S.; Lee, C.M. Treatment response and evolution of HBV resistance during lamivudine plus adefovir or entecavir therapy in patients with adefovir-resistant mutants. Antivir. Ther. 2012, 17, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, C.M.; Cheng, J.; Liang, Z.L.; Zhong, Y.W.; Ren, X.Q.; Xu, Z.H.; Zoulim, F.; Xu, D.P. Hepatitis B virus in tenofovir-naive Chinese patients with chronic hepatitis B contains no mutation of rtA194T conferring a reduced tenofovir susceptibility. Chin. Med. J. Engl. 2009, 122, 1585–1586. [Google Scholar]
- Özgüler, M.; Sayan, M. Could resistant and escape variants of hepatitis B virus be a problem in the future? Future Virol. 2018, 13, 171–179. [Google Scholar] [CrossRef]
- Romanò, L.; Paladini, S.; Galli, C.; Raimondo, G.; Pollicino, T.; Zanetti, A.R. Hepatitis B vaccination: Are escape mutant viruses a matter of concern? Hum. Vaccines Immunother. 2015, 11, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163. [Google Scholar] [CrossRef]
- Liang, M.; Ma, S.; Hu, X. Cellular immune responses in patients with hepatitis B surface antigen seroclearance induced by antiviral therapy. Virol. J. 2011, 8, 69. [Google Scholar] [CrossRef]
- Den Brouw, M.L.; Binda, R.S.; van Roosmalen, M.H.; Protzer, U.; Janssen, H.L.A.; van der Molen, R.G.; Woltman, A.M. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: A possible immune escape mechanism of hepatitis B virus. Immunology 2009, 126, 280–289. [Google Scholar] [CrossRef]
- Hamadalnil, Y.M.; Bakheit, S. Hepatitis B virus_surface gene mutations and their clinical implications. Sudan J. Med. Sci. 2017, 12, 101–113. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Primers | Sequence (5′ → 3′) | Direction | Nucleotide |
|---|---|---|---|
| 1st Round PCR | |||
| Outer forward | TTTCACCTCTGCCTAATCATCT | Forward | 1823 |
| Outer reverse | CAGACCAATTTATGCCTACAGCCT | Reverse | 1801 |
| 2nd Round PCR | |||
| Forward (F1) | GGTCACCATATTCTTGGGAAC | Forward | 2821 |
| Reverse (R1) | TGAGAGAAGTCCACCACGAGT | Reverse | 272 |
| Forward (F2) | CTAGGACCCCTGCTCGTGTT | Forward | 179 |
| Reverse (R2) | CGAACCACTGAACAAATGGCACT | Reverse | 704 |
| Forward (F3) | GTATTCCCATCCCATCATCCTG | Forward | 599 |
| Reverse (R3) | GCTAGGAGTTCCGCAGTATGG | Reverse | 1286 |
| Gender | Age Range | Therapy (No of Patients) | ALT | AST | Viral Load IU/mL |
|---|---|---|---|---|---|
| Female 14 (35%) Male 26 (65%) | 10–65 | Entecavir (09) Lamivudine + tenofovir (11) Lamivudine + entecavir (6) Tenofovir (08) Adefovir (06) | Mean ± SD 73.77 ± 10.43 Median 73.12 Range 56–95 | Mean 71.03 ± 9.10 Median 70.52 Range 52–93 | 3.0 × 104 to 5.6 × 107 |
| S. No | Drug Resistance Mutations | Therapy Used | Small Surface Gena Mutation |
|---|---|---|---|
| 1 | L180M, S202G, M204V | Lamivudine + entecavir | sI195M, sS193L |
| 2 | L180M, S202G, M204V | Lamivudine + entecavir | sI195M, A166V/T |
| 3 | L180M, S202G, M204V | Entecavir | sI195M |
| 4 | Y135S, V173M, L180M, S202G, M204V, N248H | entecavir | sI195M |
| 5 | S202G, M204V, L180M | Lamivudine+ entecavir | sI195M, A166V/T, Q129H (Vaccine escape mutation), Q181K/R |
| 6 | rtM204V + L180M, S202G | Entecavir | Not detected |
| 7 | rtM204V + L180M | Lamivudine + entecavir | sI195M, Q181K/R |
| 8 | rtM204V + L180M, V173L/M | Lamivudine + entecavir | sI195M, Q181K/R |
| 9 | rtM204V + L180M | Lamivudine + entecavir | sI195M |
| 10 | T184A | Entecavir | Not detected |
| 11 | A194V | Tenofovir | Not detected |
| 12 | Not detected | Tenofovir | Not detected |
| 13 | Q215P, V191G | Lamivudine + Tenofovir | P203S/R, P142L (vaccine escape mutation), W156C, P203R |
| 14 | N236T | Adefovir | Not detected |
| 15 | N238H/T | Tenofovir | Not detected |
| 16 | Not detected | Adefovir | Not detected |
| 17 | Not detected | Lamivudine + tenofovir | P153S, W156C, P203R |
| 18 | T184A | Entecavir | Not detected |
| 19 | Not detected | Lamivudine + tenofovir | A166V/T |
| 20 | Not detected | Tenofovir | Not detected |
| 21 | T184A | Entecavir | Not detected |
| 22 | Not detected | Tenofovir | Not detected |
| 23 | V191G | Lamivudine + tenofovir | A166V/T, V184R, A17E |
| 24 | Not detected | Tenofovir | Not detected |
| 25 | Not detected | Entecavir | Not detected |
| 26 | Not detected | Adefovir | Not detected |
| 27 | Not detected | Lamivudine + tenofovir | P153S |
| 28 | N236T | Adefovir | S193L, S132F, V184R |
| 29 | Not detected | Lamivudine + tenofovir | A166V/T, Q181K/R |
| 30 | Not detected | Tenofovir | Not detected |
| 31 | Not detected | entecavir | P203R, C76Y, Q129H (vaccine escape mutation), P142L, Q181K/R |
| 32 | T184A | Entecavir | Not detected |
| 33 | Not detected | Lamivudine + tenofovir | Not detected |
| 34 | Not detected | Lamivudine + tenofovir | Not detected |
| 35 | Not detected | Tenofovir | Not detected |
| 36 | Not detected | Lamivudine + tenofovir | A166V/T, V184R |
| 37 | Not detected | Adefovir | Not detected |
| 38 | Not detected | Lamivudine + tenofovir | Not detected |
| 39 | N238H/T | Adefovir | Not detected |
| 40 | Not detected | Lamivudine + tenofovir | A166V/T, A17E |
| Amino Acid Substitution | Frequency | Percentage % |
|---|---|---|
| N248H | 37 | 92.5 |
| N459Y | 14 | 35 |
| N131D | 8 | 20 |
| P130Q | 7 | 17.5 |
| S189Q | 5 | 12.5 |
| Y257H | 4 | 10 |
| C262S | 4 | 10 |
| V190M | 3 | 7.5 |
| L145M | 2 | 5 |
| P161S | 2 | 5 |
| V253I | 2 | 5 |
| E271D | 2 | 5 |
| A223S | 1 | 2.5 |
| L231S/W | 1 | 2.5 |
| T322S | 1 | 2.5 |
| Other Polymerase Gene Mutation | Frequency | Percentage % |
|---|---|---|
| S709L/R | 10 | 25 |
| C256 | 10 | 25 |
| E718K | 5 | 12.5 |
| M699I | 3 | 7.5 |
| T707P | 2 | 5 |
| L712 | 1 | 2.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gohar, M.; Rehman, I.U.; Ullah, A.; Khan, M.A.; Yasmin, H.; Ahmad, J.; Butt, S.; Ahmad, A. Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients. Microorganisms 2023, 11, 2622. https://doi.org/10.3390/microorganisms11112622
Gohar M, Rehman IU, Ullah A, Khan MA, Yasmin H, Ahmad J, Butt S, Ahmad A. Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients. Microorganisms. 2023; 11(11):2622. https://doi.org/10.3390/microorganisms11112622
Chicago/Turabian StyleGohar, Maryam, Irshad Ur Rehman, Amin Ullah, Muhammad Ajmal Khan, Humaira Yasmin, Jamshaid Ahmad, Sadia Butt, and Ajaz Ahmad. 2023. "Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients" Microorganisms 11, no. 11: 2622. https://doi.org/10.3390/microorganisms11112622