
Effect of Site and Phenological Status on the Potato Bacterial Rhizomicrobiota

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Potato Field Selection, Sampling, and Physicochemical Analysis
2.2. eDNA Extraction and 16S rRNA Gene Amplicon Sequencing
2.3. Raw Data Processing, Clustering, and Taxonomic Assignment of Reads
2.4. Diversity of the Rhizosphere Microbiota
2.5. Effect of Site and Phenological Stage on Rhizosphere Taxonomic Composition
2.6. Functional Inference on the Rhizosphere Microbiota
3. Results
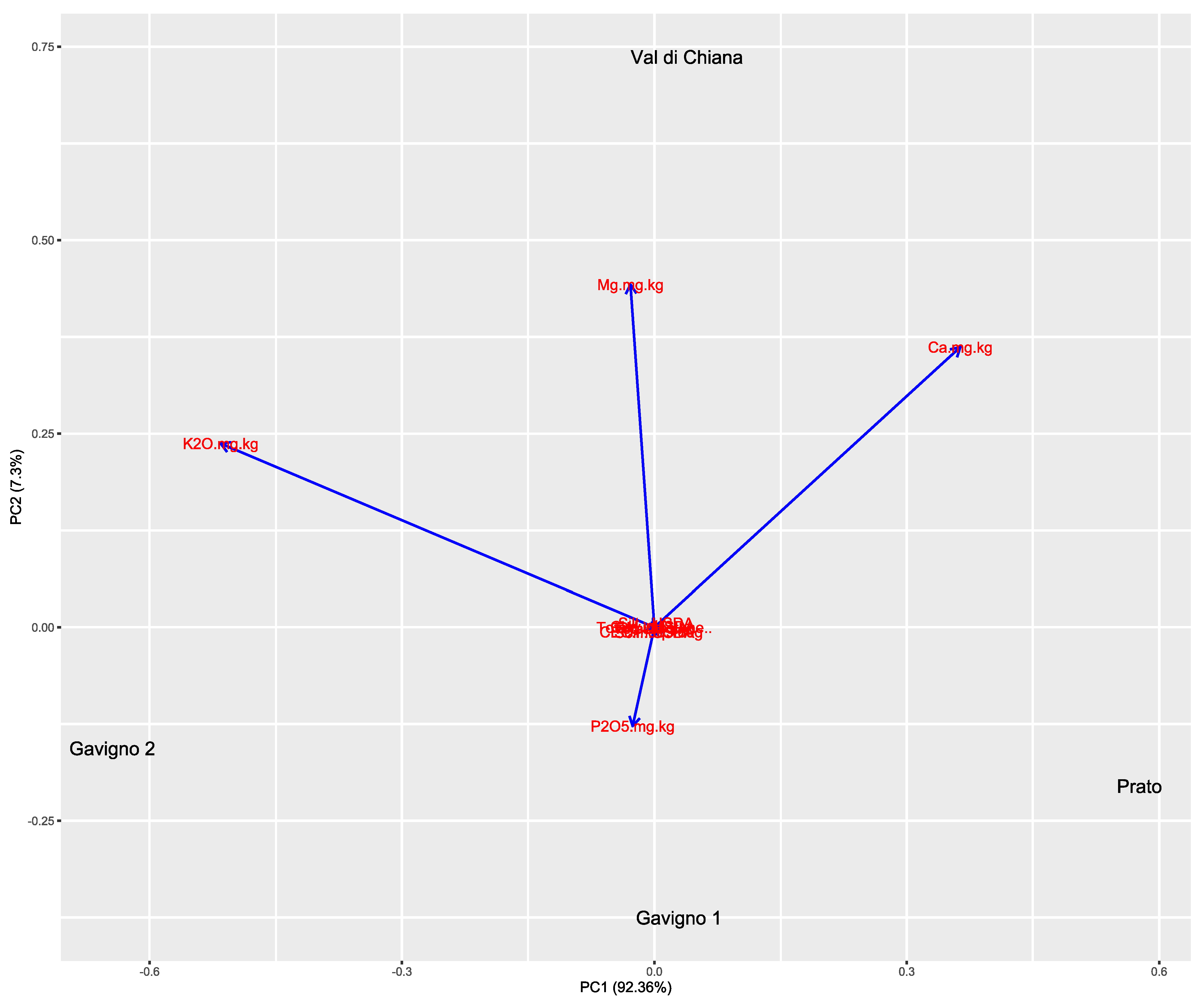
3.1. Pedoclimatic Conditions
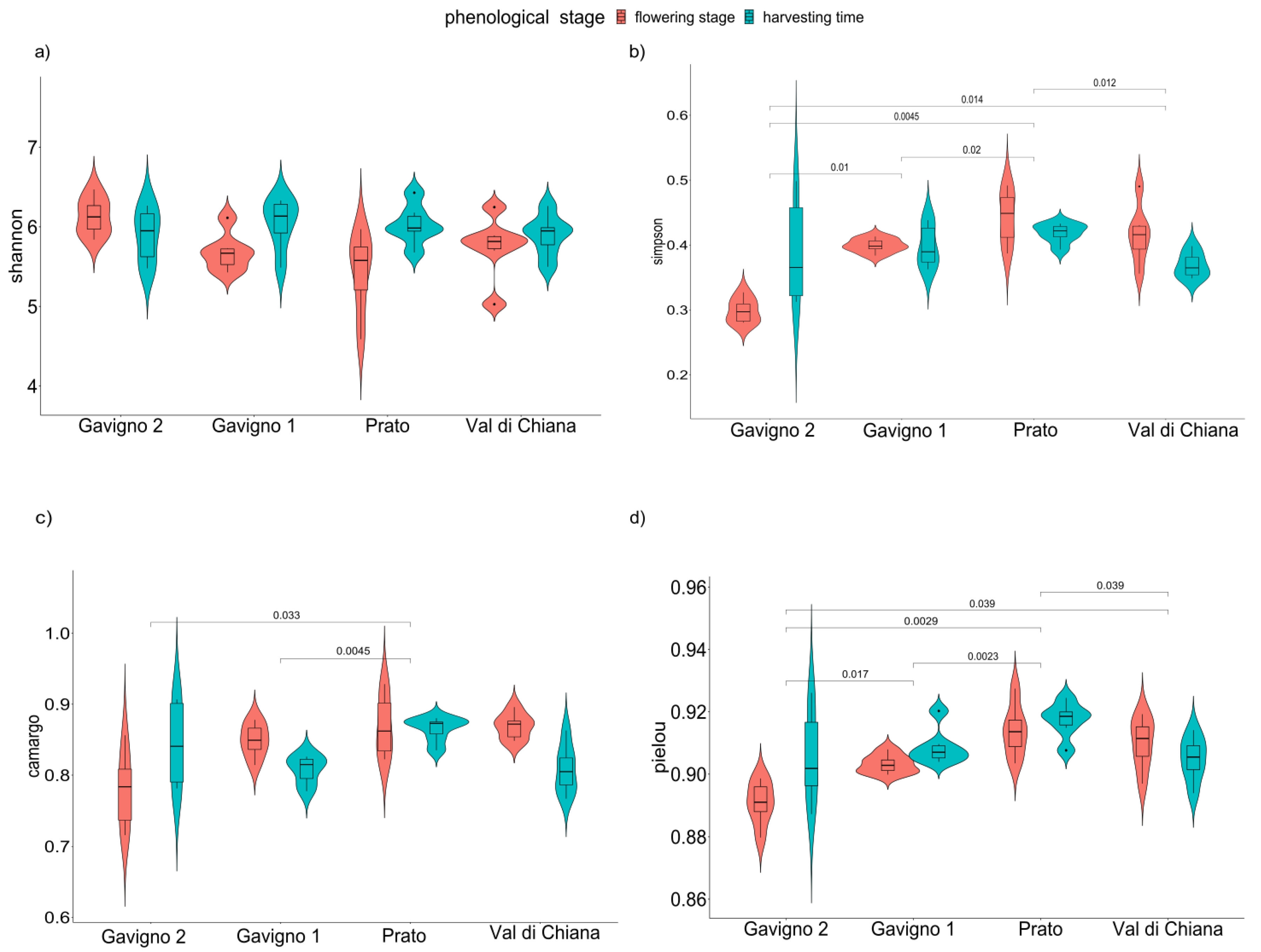
3.2. Overall Diversity of the Rhizosphere Bacterial Microbiota
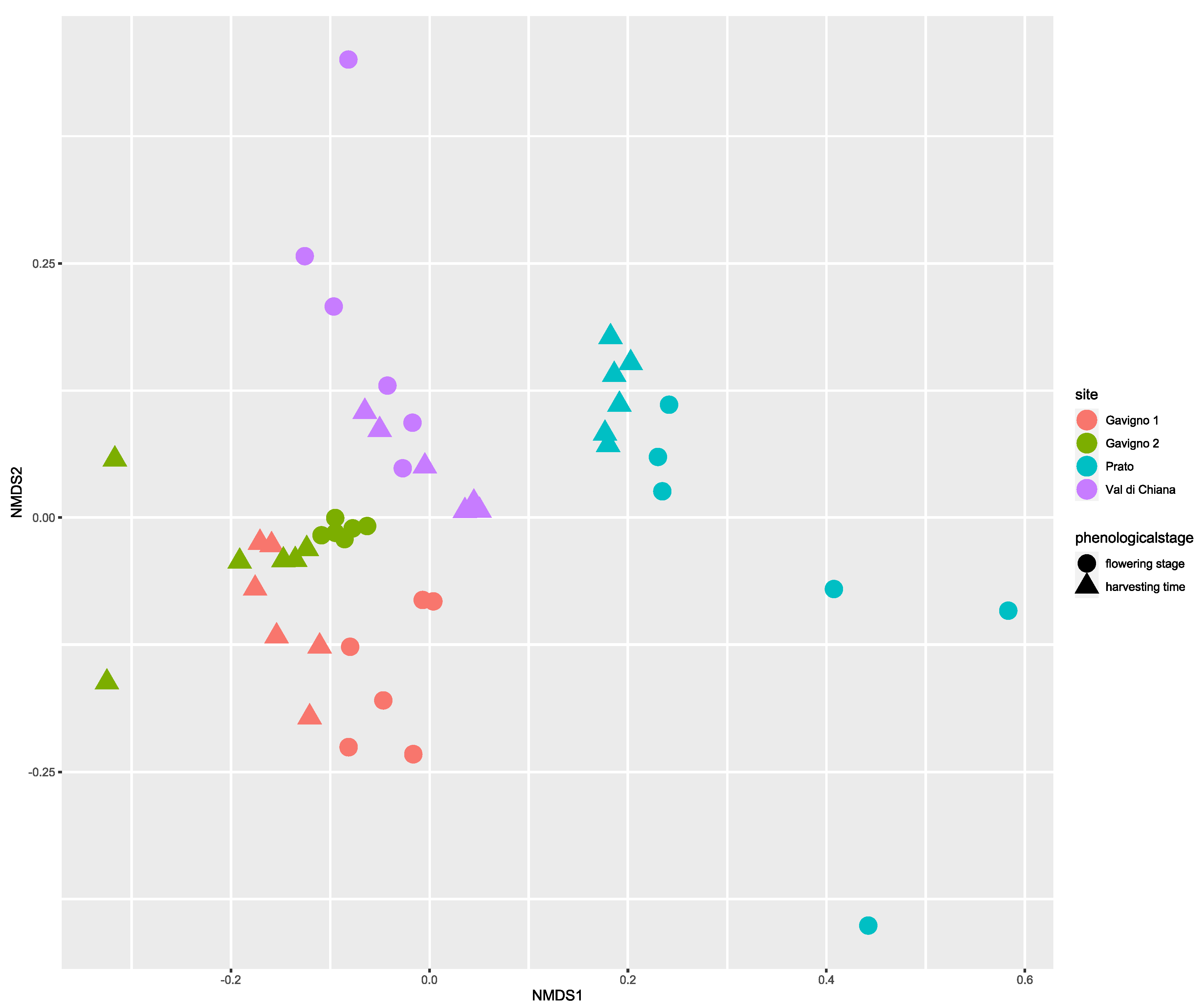
3.3. Location Is the Main Driver of the Taxonomic Composition of Potato Bacterial Rhizomicrobiota
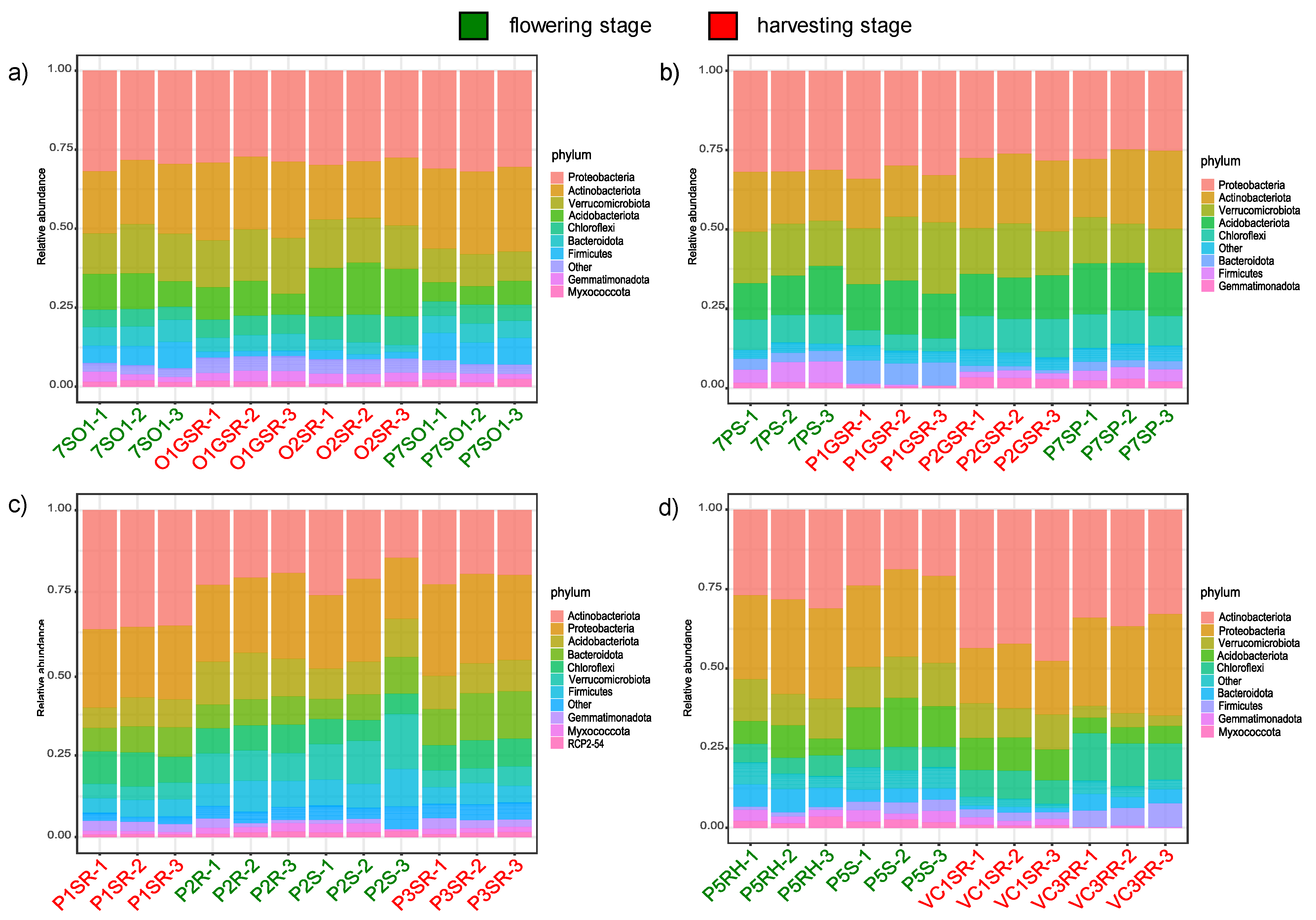
3.4. Taxonomic Signatures of Localities and Phenological Stages
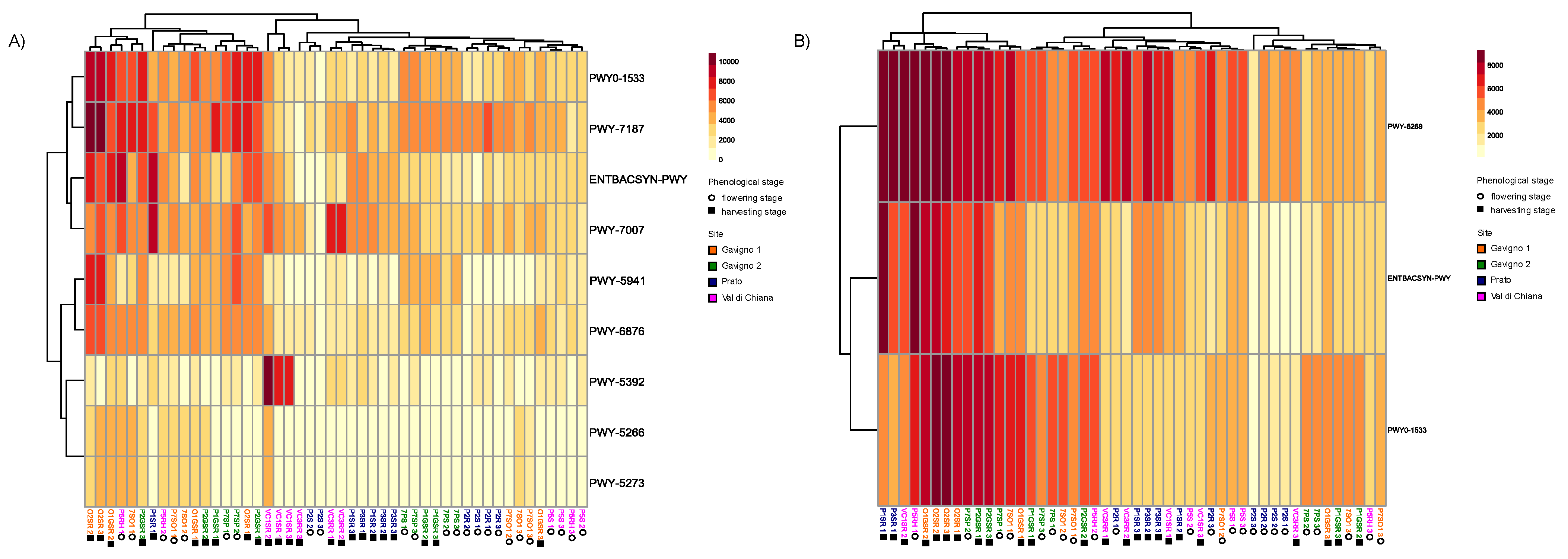
3.5. Functional Potential of the Potato Rhizomicrobiota
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motyka-Pomagruk, A.; Zoledowska, S.; Sledz, W.; Lojkowska, E. The occurrence of bacteria from different species of Pectobacteriaceae on seed potato plantations in Poland. Eur. J. Plant Pathol. 2021, 159, 309–325. [Google Scholar] [CrossRef]
- Busby, P.E.; Soman, C.; Wagner, M.R.; Friesen, M.L.; Kremer, J.; Bennett, A.; Morsy, M.; Eisen, J.A.; Leach, J.E.; Dangl, J.L. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 2017, 15, e2001793. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Bulgarelli, D. The Plant Microbiome at Work. Mol. Plant-Microbe Interact. MPMI 2015, 212, 212–217. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Sánchez-Cañizares, C.; Jorrín, B.; Poole, P.S.; Tkacz, A. Understanding the holobiont: The interdependence of plants and their microbiome. Curr. Opin. Microbiol. 2017, 38, 188–196. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.T.; Weigel, D.; Kemen, E.M. Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93, fiw242. [Google Scholar] [CrossRef]
- Hou, Q.; Wang, W.; Yang, Y.; Hu, J.; Bian, C.; Jin, L.; Li, G.; Xiong, X. Rhizosphere microbial diversity and community dynamics during potato cultivation. Eur. J. Soil Biol. 2020, 98, 103176. [Google Scholar] [CrossRef]
- Zimudzi, J.; van der Waals, J.E.; Coutinho, T.A.; Cowan, D.A.; Valverde, A. Temporal shifts of fungal communities in the rhizosphere and on tubers in potato fields. Fungal Biol. 2018, 122, 928–934. [Google Scholar] [CrossRef]
- Inceoǧlu, Ö.; Salles, J.F.; Van Overbeek, L.; Van Elsas, J.D. Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields. Appl. Environ. Microbiol. 2010, 76, 3675–3684. [Google Scholar] [CrossRef]
- Riva, V.; Mapelli, F.; Bagnasco, A.; Mengoni, A.; Borin, S.S. A Meta-Analysis Approach to De fi ning the Culturable Core of Plant Endophytic Bacterial Communities. Appl. Environ. Microbiol. 2022, 88, e02537-21. [Google Scholar] [CrossRef]
- Cangioli, L.; Vaccaro, F.; Fini, M.; Mengoni, A. Scent of a Symbiont: The Personalized Genetic Relationships of Rhizobium—Plant Interaction. Int. J. Mol. Sci. 2022, 23, 3358. [Google Scholar] [CrossRef]
- Vaccaro, F.; Cangioli, L.; Mengoni, A.; Fagorzi, C. Synthetic plant microbiota challenges in nonmodel species. Trends Microbiol. 2022. [Google Scholar] [CrossRef]
- Alegria Terrazas, R.; Balbirnie-Cumming, K.; Morris, J.; Hedley, P.E.; Russell, J.; Paterson, E.; Baggs, E.M.; Fridman, E.; Bulgarelli, D. A footprint of plant eco-geographic adaptation on the composition of the barley rhizosphere bacterial microbiota. Sci. Rep. 2020, 10, 12916. [Google Scholar] [CrossRef]
- Lin, M.; Xiong, H.; Xiang, X.; Zhou, Z.; Liang, L.; Mei, Z. The effect of plant geographical location and developmental stage on root-associated microbiomes of Gymnadenia conopsea. Front. Microbiol. 2020, 11, 1257. [Google Scholar] [CrossRef]
- Bhagat, N.; Sharma, S.; Ambardar, S.; Raj, S.; Trakroo, D.; Horacek, M.; Zouagui, R.; Sbabou, L.; Vakhlu, J. Microbiome Fingerprint as Biomarker for Geographical Origin and Heredity in Crocus sativus: A Feasibility Study. Front. Sustain. Food Syst. 2021, 5, 688393. [Google Scholar] [CrossRef]
- FAOSTAT. Primary Crops Statistical Database; FAO: Rome, Italy, 2004. [Google Scholar]
- Menna, C.; Tosco, D.; Colantuoni, F.; Caracciolo, F.; sannino, G.; Cicia, G.; Cembalo, L.; D’amico, M.; Lombardi, P.; Maraglino, T. Country-of-origin labelling for the Italian early potato supply chain. New Medit Mediterr. J. Econ. Agric. Environ. Rev. Méditerranéenne d’Economie Agric. Environ. 2013, 12, 37. [Google Scholar]
- Ingallina, C.; Spano, M.; Sobolev, A.P.; Esposito, C.; Santarcangelo, C.; Baldi, A.; Daglia, M.; Mannina, L. Characterization of Local Products for Their Industrial Use: The Case of Italian Potato Cultivars Analyzed by Untargeted and Targeted Methodologies. Foods 2020, 9, 1216. [Google Scholar] [CrossRef]
- Manzelli, M.; Romagnoli, S.; Ghiselli, L.; Benedettelli, S.; Palchetti, E.; Andrenelli, L.; Vecchio, V. Typicity in potato: Characterization of geographic origin. Ital. J. Agron. 2010, 5, 61–68. [Google Scholar] [CrossRef]
- Pacifico, D. Upland Italian Potato Quality—A Perspective. Sustainability 2018, 10, 3939. [Google Scholar] [CrossRef]
- Arnold, C.Y. The determination and significance of the base temperature in a linear heat unit system. Proc. Am. Soc. Hortic. Sci. 1959, 74, 430–445. [Google Scholar]
- Mix, K.; Rast, W.; Lopes, V.L. Increases in growing degree days in the Alpine Desert of the San Luis Valley, Colorado. Water. Air. Soil Pollut. 2010, 205, 289–304. [Google Scholar] [CrossRef]
- Singh, B.K.; Trivedi, P.; Egidi, E.; Macdonald, C.A.; Delgado-Baquerizo, M. Crop microbiome and sustainable agriculture. Nat. Rev. Microbiol. 2020, 18, 601–602. [Google Scholar] [CrossRef]
- Cangioli, L.; Mancini, M.; Napoli, M.; Fagorzi, C.; Orlandini, S.; Vaccaro, F. Differential Response of Wheat Rhizosphere Bacterial Community to Plant Variety and Fertilization. Int. J. Mol. Sci. 2022, 23, 3616. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Checcucci, A.; Maida, I.; Bacci, G.; Ninno, C.; Bilia, A.R.; Biffi, S.; Firenzuoli, F.; Flamini, G.; Fani, R.; Mengoni, A. Is the plant-associated microbiota of Thymus spp. adapted to plant essential oil? Res. Microbiol. 2017, 168, 276–282. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Wright, E.S.; Yilmaz, L.S.; Noguera, D.R. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 2012, 78, 717–725. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.A.; Lahti, L. Microbiome data science. J. Biosci. 2019, 44, 115. [Google Scholar] [CrossRef]
- Camargo, J.A. On Measuring Species Evenness and Other Associated Parameters of Community Structure. Oikos 1995, 74, 538–542. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Wickham Hadley. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Kandlikar, G.S.; Gold, Z.J.; Cowen, M.C.; Meyer, R.S.; Freise, A.C.; Kraft, N.J.B.; Moberg-Parker, J.; Sprague, J.; Kushner, D.J.; Curd, E.E. ranacapa: An R package and Shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Research 2018, 7, 1734. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Feng, K.; Zhang, Z.; Cai, W.; Liu, W.; Xu, M.; Yin, H.; Wang, A.; He, Z.; Deng, Y. Biodiversity and species competition regulate the resilience of microbial biofilm community. Mol. Ecol. 2017, 26, 6170–6182. [Google Scholar] [CrossRef]
- Staff, S.S.L. Soil Survey Laboratory Methods Manual; National Soil Survey Center: Lincoln, NE, USA, 1996. [Google Scholar]
- Buchholz, F.; Antonielli, L.; Kostić, T.; Sessitsch, A.; Mitter, B. The bacterial community in potato is recruited from soil and partly inherited across generations. PLoS ONE 2019, 14, e0223691. [Google Scholar] [CrossRef]
- Liu, J.; Nothias, L.-F.; Dorrestein, P.C.; Tahlan, K.; Bignell, D.R.D. Genomic and Metabolomic Analysis of the Potato Common Scab Pathogen Streptomyces scabiei. ACS Omega 2021, 6, 11474–11487. [Google Scholar] [CrossRef]
- Hiltunen, L.H.; Tarvainen, O.; Kelloniemi, J.; Tanskanen, J.; Karhu, J.; Valkonen, J.P.T. Soil bacterial community in potato tuberosphere following repeated applications of a common scab suppressive antagonist. Appl. Soil Ecol. 2021, 167, 104096. [Google Scholar] [CrossRef]
- Qi, B.; Han, M. Microbial Siderophore Enterobactin Promotes Mitochondrial Iron Uptake and Development of the Host via Interaction with ATP Synthase. Cell 2018, 175, 571–582.e11. [Google Scholar] [CrossRef] [Green Version]
- Ling-yun, H.; Kyle, W.D.; Helene, A.-P.; Michael, M.; Barak, J.D. Requirement of Siderophore Biosynthesis for Plant Colonization by Salmonella enterica. Appl. Environ. Microbiol. 2012, 78, 4561–4570. [Google Scholar] [CrossRef]
- Vandana, U.K.; Rajkumari, J.; Singha, L.P.; Satish, L.; Alavilli, H.; Sudheer, P.D.V.N.; Chauhan, S.; Ratnala, R.; Satturu, V.; Mazumder, P.B.; et al. The Endophytic Microbiome as a Hotspot of Synergistic Interactions, with Prospects of Plant Growth Promotion. Biology 2021, 10, 101. [Google Scholar] [CrossRef]
- Oleńska, E.; Małek, W.; Wójcik, M.; Swiecicka, I.; Thijs, S.; Vangronsveld, J. Beneficial features of plant growth-promoting rhizobacteria for improving plant growth and health in challenging conditions: A methodical review. Sci. Total Environ. 2020, 743, 140682. [Google Scholar] [CrossRef]
- Perkowska, I.; Potrykus, M.; Siwinska, J.; Siudem, D.; Lojkowska, E.; Ihnatowicz, A. Interplay between coumarin accumulation, iron deficiency and plant resistance to Dickeya spp. Int. J. Mol. Sci. 2021, 22, 6449. [Google Scholar] [CrossRef]
- Stassen, M.J.J.; Hsu, S.H.; Pieterse, C.M.J.; Stringlis, I.A. Coumarin Communication Along the Microbiome–Root–Shoot Axis. Trends Plant Sci. 2021, 26, 169–183. [Google Scholar] [CrossRef]
- Pattnaik, S.; Mohapatra, B.; Gupta, A. Plant Growth-Promoting Microbe Mediated Uptake of Essential Nutrients (Fe, P, K) for Crop Stress Management: Microbe–Soil–Plant Continuum. Front. Agron. 2021, 58, 689972. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.; Wang, J.; Singh, B.; Han, L.; Shen, J.; Li, P.; Wang, G.; Wu, C.; Ge, A.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2020, 229, 1091–1104. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Abedini, D.; Jaupitre, S.; Bouwmeester, H.; Dong, L. Metabolic interactions in beneficial microbe recruitment by plants. Curr. Opin. Biotechnol. 2021, 70, 241–247. [Google Scholar] [CrossRef]
- diCenzo, G.C.; Tesi, M.; Pfau, T.; Mengoni, A.; Fondi, M. Genome-scale metabolic reconstruction of the symbiosis between a leguminous plant and a nitrogen-fixing bacterium. Nat. Commun. 2020, 11, 2574. [Google Scholar] [CrossRef]
- Giupponi, L.; Leoni, V.; Pedrali, D.; Ceciliani, G.; Bassoli, A.; Borgonovo, G. Morphometric and Phytochemical Characterization and Elevation Effect on Yield of Three Potato Landraces of the Ligurian Apennines (Northern Italy). J. Appl. Bot. Food Qual. 2020, 93, 234–243. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Geographical Coordinates | Altitude | NPK Fertilization Rate |
|---|---|---|---|
| Gavigno 1 | 44.068862° N, 11.093375° E | 739 m | 15-5-5 (500 g ha−1) |
| Gavigno 2 | 44.062105° N, 11.095891° E | 763 m | 15-5-5 (500 kg ha−1) |
| Val di Chiana | 43.412705° N, 11.784870° E | 260 m | 15-9-15 (100 kg ha−1) |
| Prato | 43.860192° N, 11.054822° E | 39 m | 12-12-17 (350 kg ha−1) |
| Shannon | Simpson | Pielou | Camargo | |||||
|---|---|---|---|---|---|---|---|---|
| F | p-Value | F | p-Value | F | p-Value | F | p-Value | |
| Locality | 15.064 | n.s. | 11.9274 | 0.001 | 6.2539 | 0.004 | 4.4182 | 0.011 |
| Stage | 52.131 | 0.018 | 0.4555 | n.s. | 2.7498 | n.s. | 0.6968 | n.s. |
| Locality:stage | 32.784 | 0.03 | 7.5625 | 0.002 | 4.7729 | 0.008 | 6.7443 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cangioli, L.; Mancini, M.; Baldi, A.; Fagorzi, C.; Orlandini, S.; Vaccaro, F.; Mengoni, A. Effect of Site and Phenological Status on the Potato Bacterial Rhizomicrobiota. Microorganisms 2022, 10, 1743. https://doi.org/10.3390/microorganisms10091743
Cangioli L, Mancini M, Baldi A, Fagorzi C, Orlandini S, Vaccaro F, Mengoni A. Effect of Site and Phenological Status on the Potato Bacterial Rhizomicrobiota. Microorganisms. 2022; 10(9):1743. https://doi.org/10.3390/microorganisms10091743
Chicago/Turabian StyleCangioli, Lisa, Marco Mancini, Ada Baldi, Camilla Fagorzi, Simone Orlandini, Francesca Vaccaro, and Alessio Mengoni. 2022. "Effect of Site and Phenological Status on the Potato Bacterial Rhizomicrobiota" Microorganisms 10, no. 9: 1743. https://doi.org/10.3390/microorganisms10091743