
Biochemical and Kinetic Characterization of the Glucose-6-Phosphate Dehydrogenase from Helicobacter pylori Strain 29CaP

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Plasmids
2.2. Reagents
2.3. Alignment of Sequences
2.4. Construction of pET3aHisTEVP-zwf Vector
2.5. Expression and Purification of the G6PD Protein from Helicobacter pylori
2.6. Enzymatic Activity Assay
2.7. Oligomeric Composition Analysis by Molecular Exclusion
2.8. Determination of the Functional Parameters of the G6PD Protein of Helicobacter pylori
2.8.1. Enzyme Stability, pH, and Optimal Temperature
2.8.2. Determination of Kinetic Parameters
2.9. Spectroscopic Characterization
2.9.1. Far-UV Circular Dichroism
2.9.2. Thermal Stability, Monitored by Circular Dichroism
2.10. Evaluation of the Stability of the Recombinant HpG6PD Protein
2.10.1. Stability Analysis of HpG6PD Enzyme Activity
2.10.2. Fluorescence Spectroscopy
3. Results and Discussion
3.1. Alignment of G6PDs Proteins
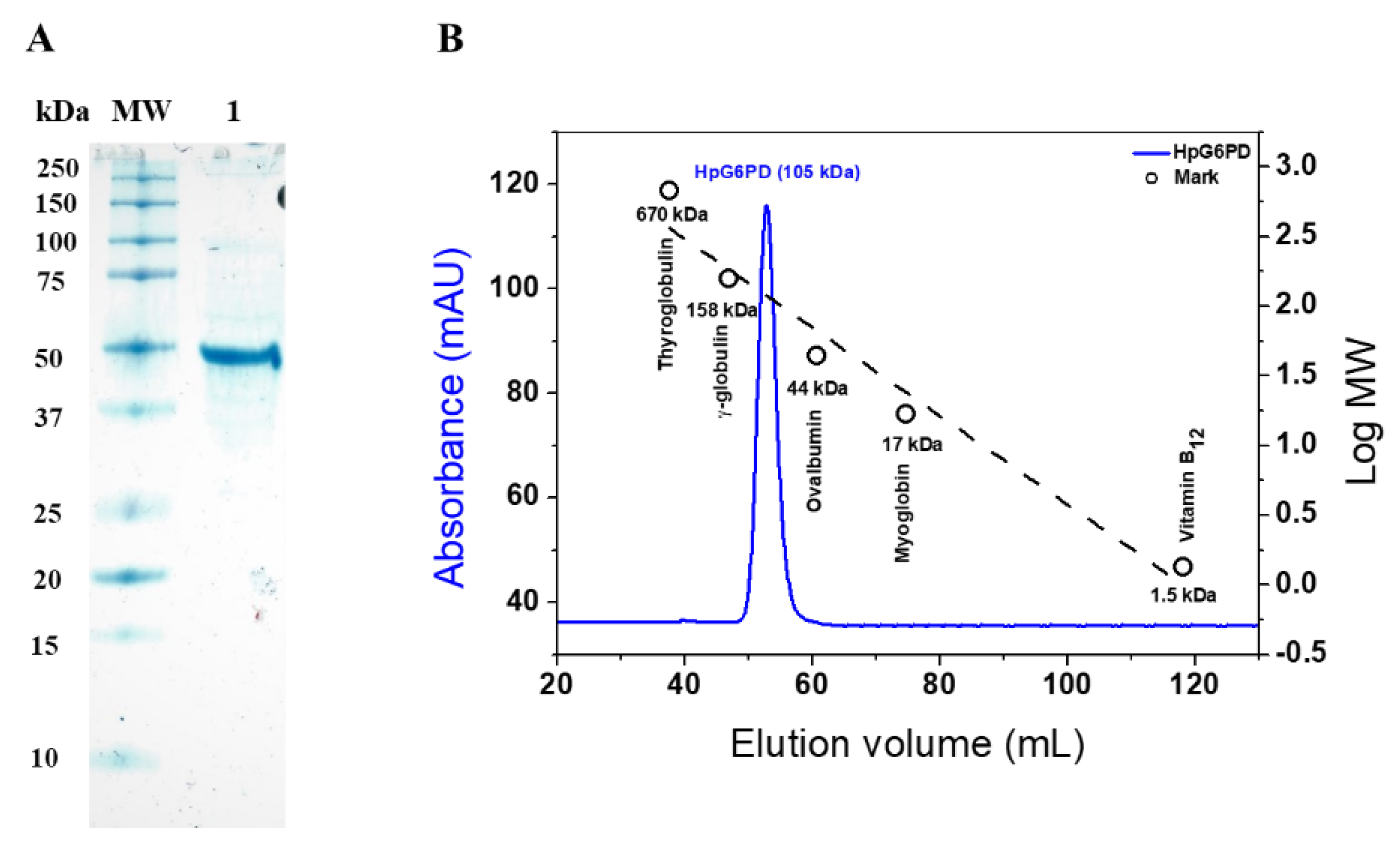
3.2. Overexpression and Purification of Recombinant HpG6PD
3.3. Determination of the Oligomeric State of the Recombinant HpG6PD
3.4. Determination of the Functional Parameters of the HpG6PD Protein
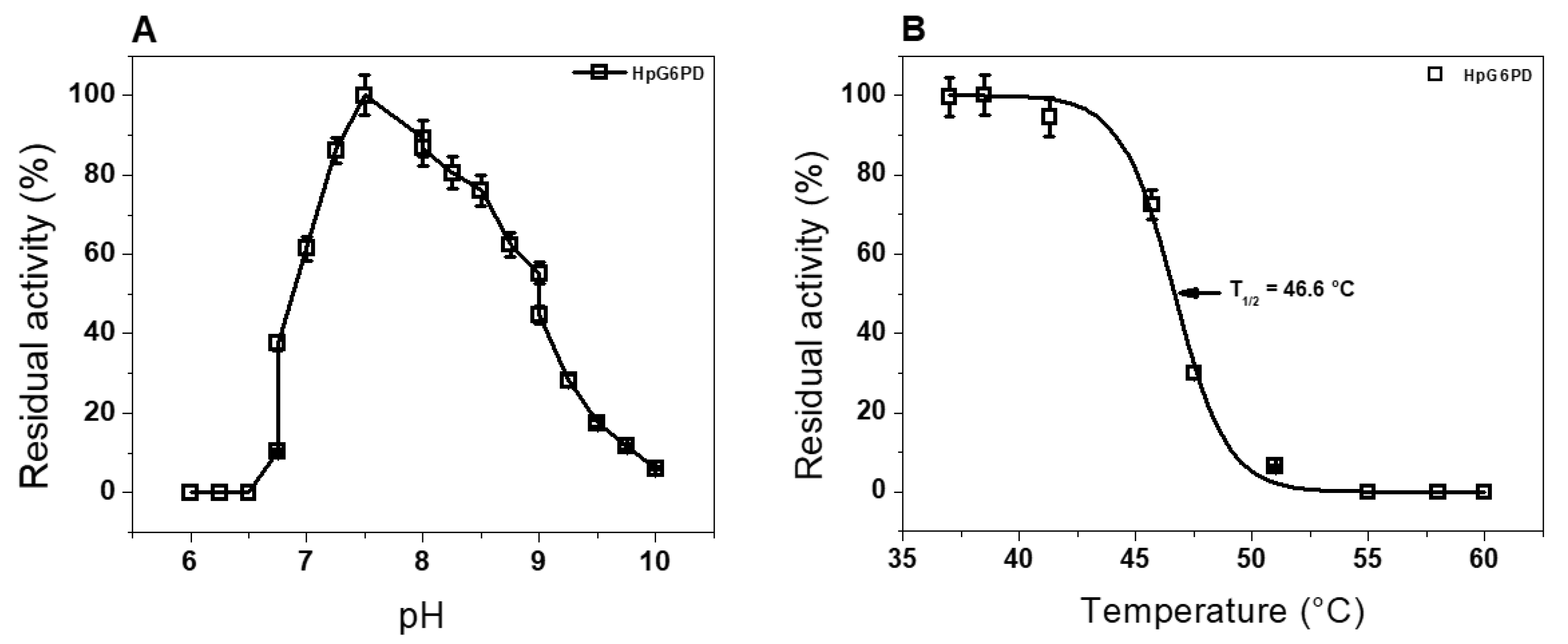
3.4.1. Effect of pH and Temperature Stability
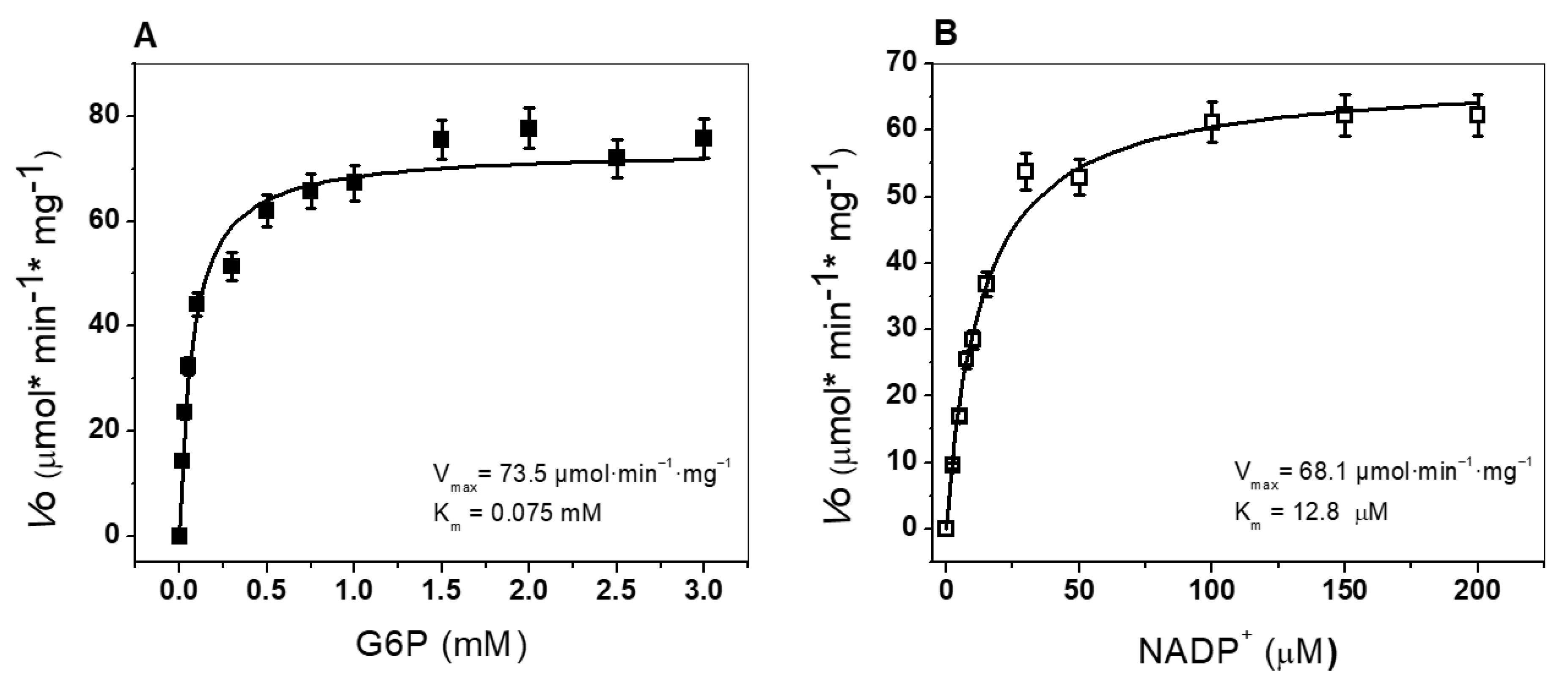
3.4.2. Determination of Kinetic Parameters
3.5. Spectroscopic Characterization
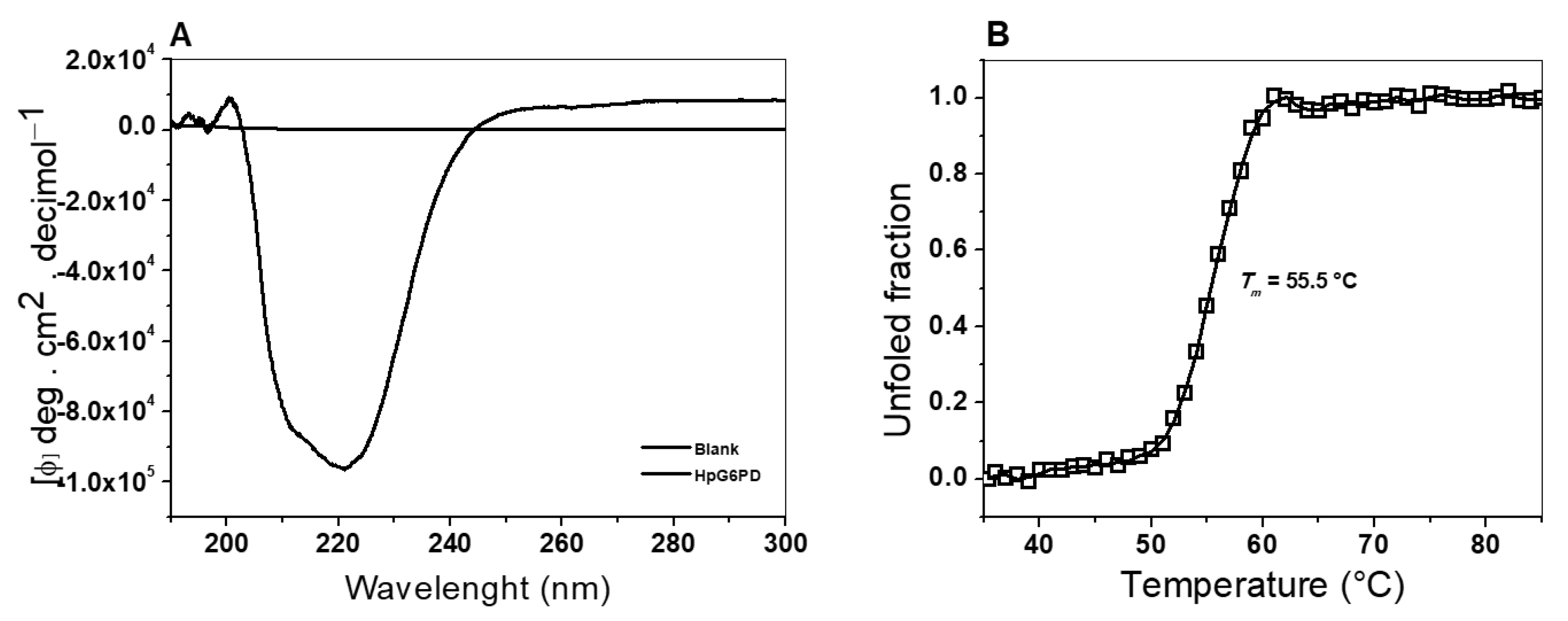
3.5.1. Circular Dichroism (CD) Assay
3.5.2. Thermal Stability Assay, Followed by CD
3.6. Evaluation of the Stability of the Recombinant HpG6PD Protein
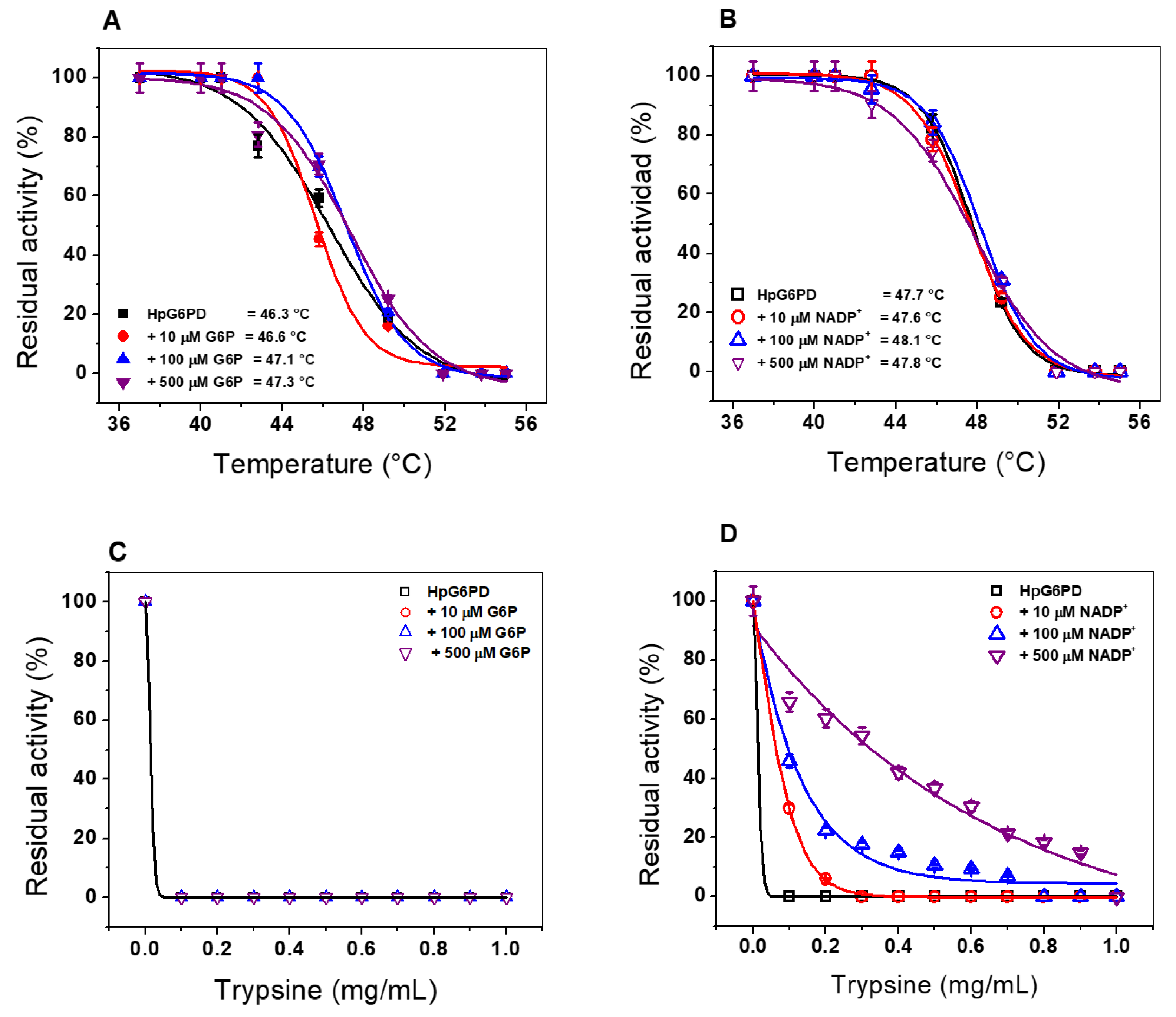
3.6.1. Thermal Inactivation Assay
3.6.2. Susceptibility of the HpG6PD Enzyme to Trypsin Proteolysis
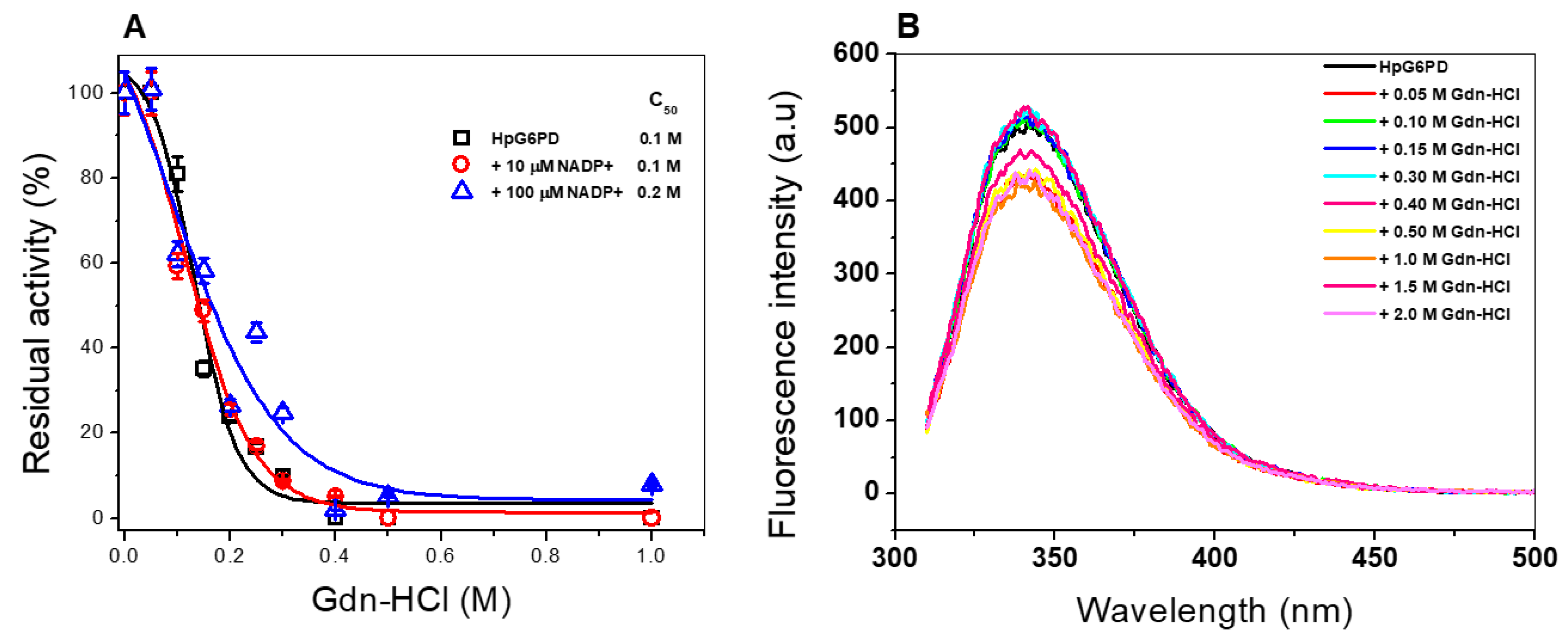
3.6.3. Stability of the HpG6PD Protein in the Presence of Guanidine Hydrochloride (Gdn-HCl)
3.6.4. Structural Analysis via Intrinsic Fluorescence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cover, T.L.; Blaser, M.J. Helicobacter pylori infection, a paradigm for chronic mucosal inflammation: Pathogenesis and implications for eradication and prevention. Adv. Intern. Med. 1996, 41, 85–117. [Google Scholar] [PubMed]
- Goodwin, C.S.; Armstrong, J.A.; Marshall, B.J. Campylobacter pyloridis, gastritis, and peptic ulceration. J. Clin. Pathol. 1986, 39, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, M.J. Not all Helicobacter pylori strains are created equal: Should all be eliminated? Lancet 1997, 349, 1020–1022. [Google Scholar] [CrossRef]
- Goodwin, C.S. Helicobacter pylori gastritis, peptic ulcer, and gastric cancer: Clinical and molecular aspects. Clin. Infect. Dis. 1997, 25, 1017–1019. [Google Scholar] [CrossRef] [Green Version]
- Blaser, M.J.; Berg, D.E. Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Investig. 2001, 107, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Kavermann, H.; Burns, B.P.; Angermuller, K.; Odenbreit, S.; Fischer, W.; Melchers, K.; Haas, R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 2003, 197, 813–822. [Google Scholar] [CrossRef]
- Kusters, J.G.; Van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Denic, M.; Touati, E.; De Reuse, H. Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2020, 25 (Suppl. S1), e12736. [Google Scholar] [CrossRef]
- Leja, M.; Grinberga-Derica, I.; Bilgilier, C.; Steininger, C. Review: Epidemiology of Helicobacter pylori infection. Helicobacter 2019, 24 (Suppl. S1), e12635. [Google Scholar] [CrossRef] [Green Version]
- Gold, B.D. Helicobacter pylori infection in children. Curr. Probl. Pediatr. Adolesc. Health Care 2001, 31, 247–266. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, Liver Flukes and Helicobacter Pylori; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1994; Volume 61. [Google Scholar]
- Yang, J.C.; Lu, C.W.; Lin, C.J. Treatment of Helicobacter pylori infection: Current status and future concepts. World J. Gastroenterol. 2014, 20, 5283–5293. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhu, Y.; Lu, N.H. Novel and Effective Therapeutic Regimens for Helicobacter pylori in an Era of Increasing Antibiotic Resistance. Front. Cell Infect. Microbiol. 2017, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Esaki, M.; Kusano, C.; Ikehara, H.; Gotoda, T. Development of Helicobacter pylori treatment: How do we manage antimicrobial resistance? World J. Gastroenterol. 2019, 25, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Chen, F. Identifying targets for drug discovery using bioinformatics. Expert Opin. Ther. Targets 2008, 12, 383–389. [Google Scholar] [CrossRef]
- Nwaka, S.; Ridley, R.G. Virtual drug discovery and development for neglected diseases through public-private partnerships. Nat. Rev. Drug Discov. 2003, 2, 919–928. [Google Scholar] [CrossRef]
- Mandal, S.; Moudgil, M.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef]
- Hazell, S.L.; Mendz, G.L. How Helicobacter pylori works: An overview of the metabolism of Helicobacter pylori. Helicobacter 1997, 2, 1–12. [Google Scholar] [CrossRef]
- Marais, A.; Mendz, G.L.; Hazell, S.L.; Megraud, F. Metabolism and genetics of Helicobacter pylori: The genome era. Microbiol. Mol. Biol. Rev. 1999, 63, 642–674. [Google Scholar] [CrossRef] [Green Version]
- Mendz, G.L.; Hazell, S.L.; Burns, B.P. Glucose utilization and lactate production by Helicobacter pylori. J. Gen. Microbiol. 1993, 139, 3023–3028. [Google Scholar] [CrossRef] [Green Version]
- Mendz, G.L.; Hazell, S.L.; Burns, B.P. The Entner-Doudoroff pathway in Helicobacter pylori. Arch. Biochem. Biophys. 1994, 312, 349–356. [Google Scholar] [CrossRef]
- Chalk, P.A.; Roberts, A.D.; Blows, W.M. Metabolism of pyruvate and glucose by intact cells of Helicobacter pylori studied by 13C NMR spectroscopy. Microbiology 1994, 140, 2085–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendz, G.L.; Burns, B.P.; Hazell, S.L. Characterisation of glucose transport in Helicobacter pylori. Biochim. Biophys. Acta 1995, 1244, 269–276. [Google Scholar] [CrossRef]
- Psakis, G.; Saidijam, M.; Shibayama, K.; Polaczek, J.; Bettaney, K.E.; Baldwin, J.M.; Baldwin, S.A.; Hope, R.; Essen, L.O.; Essenberg, R.C.; et al. The sodium-dependent D-glucose transport protein of Helicobacter pylori. Mol. Microbiol. 2009, 71, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Mucito-Varela, E.; Castillo-Rojas, G.; Cevallos, M.A.; Lozano, L.; Merino, E.; Lopez-Leal, G.; Lopez-Vidal, Y. Complete Genome Sequence of Helicobacter pylori Strain 29CaP Isolated from a Mexican Patient with Gastric Cancer. Genome Announc. 2016, 4, e01512-15. [Google Scholar] [CrossRef] [Green Version]
- Enriquez-Flores, S.; Rodriguez-Romero, A.; Hernandez-Alcantara, G.; Oria-Hernandez, J.; Gutierrez-Castrellon, P.; Perez-Hernandez, G.; De la Mora-de la Mora, I.; Castillo-Villanueva, A.; Garcia-Torres, I.; Mendez, S.T.; et al. Determining the molecular mechanism of inactivation by chemical modification of triosephosphate isomerase from the human parasite Giardia lamblia: A study for antiparasitic drug design. Proteins 2011, 79, 2711–2724. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Manzo, S.; Terron-Hernandez, J.; De la Mora-de la Mora, I.; Garcia-Torres, I.; Lopez-Velazquez, G.; Reyes-Vivas, H.; Oria-Hernandez, J. Cloning, expression, purification and characterization of his-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef]
- Ramirez-Nava, E.J.; Ortega-Cuellar, D.; Serrano-Posada, H.; Gonzalez-Valdez, A.; Vanoye-Carlo, A.; Hernandez-Ochoa, B.; Sierra-Palacios, E.; Hernandez-Pineda, J.; Rodriguez-Bustamante, E.; Arreguin-Espinosa, R.; et al. Biochemical Analysis of Two Single Mutants that Give Rise to a Polymorphic G6PD A-Double Mutant. Int. J. Mol. Sci. 2017, 18, 2244. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Gomez-Manzo, S.; Terron-Hernandez, J.; De la Mora-De la Mora, I.; Gonzalez-Valdez, A.; Marcial-Quino, J.; Garcia-Torres, I.; Vanoye-Carlo, A.; Lopez-Velazquez, G.; Hernandez-Alcantara, G.; Oria-Hernandez, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Morales, Y.Y.; Vanoye-Carlo, A.; Castillo-Rodriguez, R.A.; Serrano-Posada, H.; Gonzalez-Valdez, A.; Ortega-Cuellar, D.; Hernandez-Ochoa, B.; Moreno-Vargas, L.M.; Prada-Gracia, D.; Sierra-Palacios, E.; et al. Cloning and biochemical characterization of three glucose6phosphate dehydrogenase mutants presents in the Mexican population. Int. J. Biol. Macromol. 2018, 119, 926–936. [Google Scholar] [CrossRef]
- Ramirez-Nava, E.J.; Ortega-Cuellar, D.; Gonzalez-Valdez, A.; Castillo-Rodriguez, R.A.; Ponce-Soto, G.Y.; Hernandez-Ochoa, B.; Cardenas-Rodriguez, N.; Martinez-Rosas, V.; Morales-Luna, L.; Serrano-Posada, H.; et al. Molecular Cloning and Exploration of the Biochemical and Functional Analysis of Recombinant Glucose-6-Phosphate Dehydrogenase from Gluconoacetobacter diazotrophicus PAL5. Int. J. Mol. Sci. 2019, 20, 5279. [Google Scholar] [CrossRef] [PubMed]
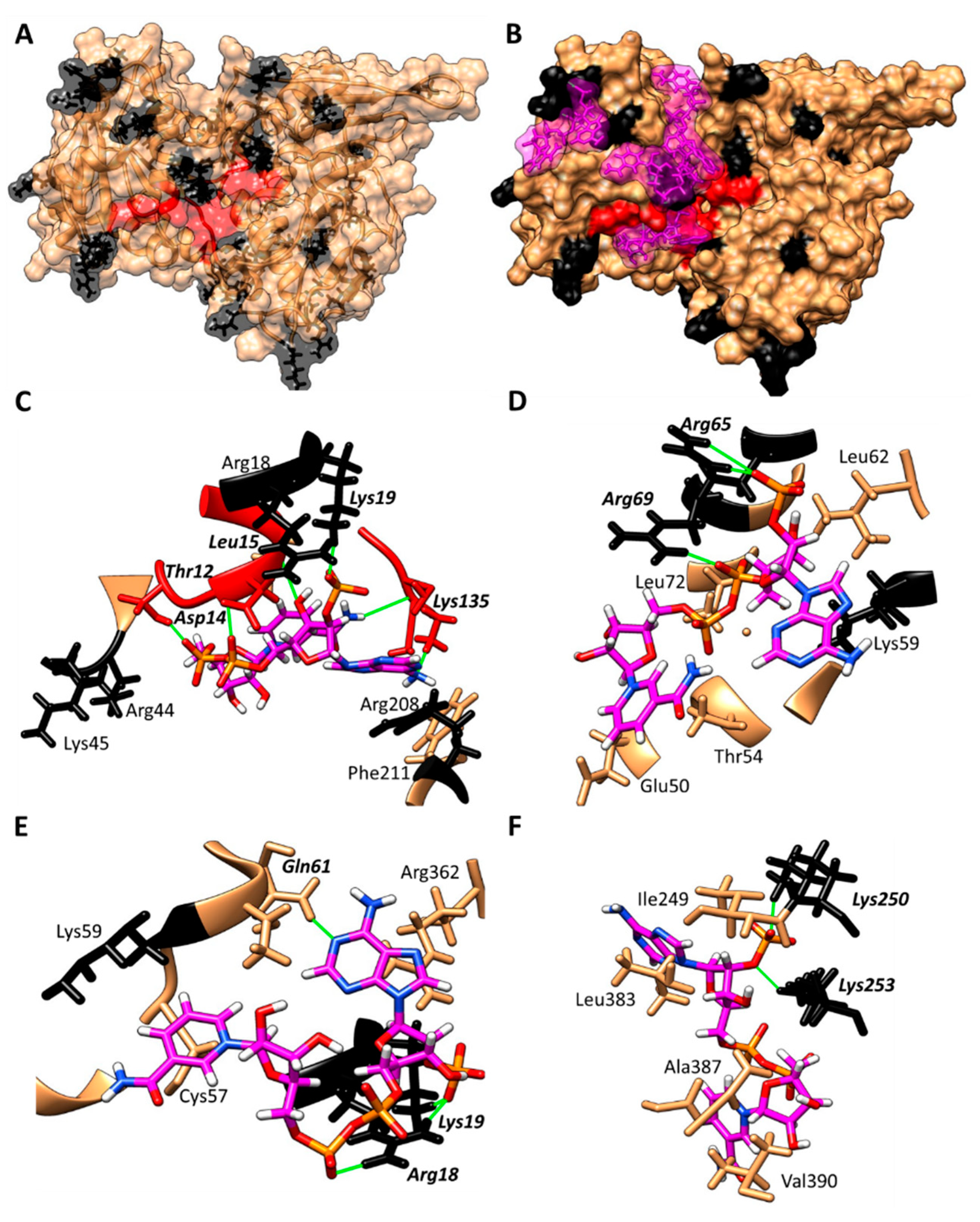
- Hernandez-Ochoa, B.; Navarrete-Vazquez, G.; Aguayo-Ortiz, R.; Ortiz-Ramirez, P.; Morales-Luna, L.; Martinez-Rosas, V.; Gonzalez-Valdez, A.; Gomez-Chavez, F.; Enriquez-Flores, S.; Wong-Baeza, C.; et al. Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors. Molecules 2021, 26, 4955. [Google Scholar] [CrossRef] [PubMed]
- Scrutton, N.S.; Berry, A.; Perham, R.N. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 1990, 343, 38–43. [Google Scholar] [CrossRef]
- Vought, V.; Ciccone, T.; Davino, M.H.; Fairbairn, L.; Lin, Y.; Cosgrove, M.S.; Adams, M.J.; Levy, H.R. Delineation of the roles of amino acids involved in the catalytic functions of Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase. Biochemistry 2000, 39, 15012–15021. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Gover, S.; Naylor, C.E.; Vandeputte-Rutten, L.; Adams, M.J.; Levy, H.R. An examination of the role of asp-177 in the His-Asp catalytic dyad of Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: X-ray structure and pH dependence of kinetic parameters of the D177N mutant enzyme. Biochemistry 2000, 39, 15002–15011. [Google Scholar] [CrossRef]
- Bautista, J.M.; Mason, P.J.; Luzzatto, L. Human glucose-6-phosphate dehydrogenase. Lysine 205 is dispensable for substrate binding but essential for catalysis. FEBS Lett. 1995, 366, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Morales-Luna, L.; Hernandez-Ochoa, B.; Ramirez-Nava, E.J.; Martinez-Rosas, V.; Ortiz-Ramirez, P.; Fernandez-Rosario, F.; Gonzalez-Valdez, A.; Cardenas-Rodriguez, N.; Serrano-Posada, H.; Centeno-Leija, S.; et al. Characterizing the Fused TvG6PD::6PGL Protein from the Protozoan Trichomonas vaginalis, and Effects of the NADP(+) Molecule on Enzyme Stability. Int. J. Mol. Sci. 2020, 21, 4831. [Google Scholar] [CrossRef]
- Rowland, P.; Basak, A.K.; Gover, S.; Levy, H.R.; Adams, M.J. The three-dimensional structure of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 A resolution. Structure 1994, 2, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Schlichting, B.; Schonheit, P. Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: Expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol. Lett. 2002, 216, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Acero-Navarro, K.E.; Jimenez-Ramirez, M.; Villalobos, M.A.; Vargas-Martinez, R.; Perales-Vela, H.V.; Velasco-Garcia, R. Cloning, overexpression, and purification of glucose-6-phosphate dehydrogenase of Pseudomonas aeruginosa. Protein Expr. Purif. 2018, 142, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Naylor, C.E.; Gover, S.; Basak, A.K.; Cosgrove, M.S.; Levy, H.R.; Adams, M.J. NADP+ and NAD+ binding to the dual coenzyme specific enzyme Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: Different interdomain hinge angles are seen in different binary and ternary complexes. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickl, A.; Schonheit, P. The oxidative pentose phosphate pathway in the haloarchaeon Haloferax volcanii involves a novel type of glucose-6-phosphate dehydrogenase—The archaeal Zwischenferment. FEBS Lett. 2015, 589, 1105–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Purification and characterization of glucose-6-phosphate dehydrogenase from camel liver. Enzyme Res. 2014, 2014, 714054. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Biochemical characterization of buffalo liver glucose-6-phosphate dehydrogenase isoforms. Protein J. 2015, 34, 193–204. [Google Scholar] [CrossRef]
- Verma, A.; Suthar, M.K.; Doharey, P.K.; Gupta, S.; Yadav, S.; Chauhan, P.M.; Saxena, J.K. Molecular cloning and characterization of glucose-6-phosphate dehydrogenase from Brugia malayi. Parasitology 2013, 140, 897–906. [Google Scholar] [CrossRef]
- Schuurmann, J.; Quehl, P.; Lindhorst, F.; Lang, K.; Jose, J. Autodisplay of glucose-6-phosphate dehydrogenase for redox cofactor regeneration at the cell surface. Biotechnol. Bioeng. 2017, 114, 1658–1669. [Google Scholar] [CrossRef]
- TranNgoc, K.; Pham, N.; Lee, C.; Jang, S.H. Cloning, Expression, and Characterization of a Psychrophilic Glucose 6-Phosphate Dehydrogenase from Sphingomonas sp. PAMC 26621. Int. J. Mol. Sci. 2019, 20, 1362. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, C.; Moraca, F.; Medeiros, A.; Botta, M.; Hamilton, N.; Comini, M.A. Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. [Google Scholar] [CrossRef] [Green Version]
- Morales-Luna, L.; Serrano-Posada, H.; Gonzalez-Valdez, A.; Ortega-Cuellar, D.; Vanoye-Carlo, A.; Hernandez-Ochoa, B.; Sierra-Palacios, E.; Rufino-Gonzalez, Y.; Castillo-Rodriguez, R.A.; Perez de la Cruz, V.; et al. Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia. Int. J. Mol. Sci. 2018, 19, 2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jortzik, E.; Mailu, B.M.; Preuss, J.; Fischer, M.; Bode, L.; Rahlfs, S.; Becker, K. Glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase: A unique bifunctional enzyme from Plasmodium falciparum. Biochem. J. 2011, 436, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennekes, L.M.; Goosen, T.; Van den Broek, P.J.; Van den Broek, H.W. Purification and characterization of glucose-6-phosphate dehydrogenase from Aspergillus niger and Aspergillus nidulans. J. Gen. Microbiol. 1993, 139, 2793–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Han, J.; Wu, J.; Chen, H. Heteroexpression and functional characterization of glucose 6-phosphate dehydrogenase from industrial Aspergillus oryzae. J. Microbiol. Biotechnol. 2019, 29, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozer, N.; Bilgi, C.; Hamdi Ogus, I. Dog liver glucose-6-phosphate dehydrogenase: Purification and kinetic properties. Int. J. Biochem. Cell Biol. 2002, 34, 253–262. [Google Scholar] [CrossRef]
- Wierenga, R.K.; Terpstra, P.; Hol, W.G. Prediction of the occurrence of the ADP-binding beta alpha beta-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol. 1986, 187, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Gonzalez-Valdez, A.; Martinez-Rosas, V.; Hernandez-Ochoa, B.; Sierra-Palacios, E.; Castillo-Rodriguez, R.A.; Cuevas-Cruz, M.; et al. Functional and Biochemical Characterization of Three Recombinant Human Glucose-6-Phosphate Dehydrogenase Mutants: Zacatecas, Vanua-Lava and Viangchan. Int. J. Mol. Sci. 2016, 17, 787. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | kcat (s−1) | Km G6P (µM) | Km NADP+ (µM) | Reference |
|---|---|---|---|---|
| Helicobacter pylori | 70 | 75 | 12 | In this study |
| Gluconacetobacter diazotrophichus | 293,181 | 63 | 7 | [34] |
| Escherichia coli DH5α | 32 | 224 | 127 | [50] |
| Pseudomonas aeruginosa | 540 | 498 | 56 | [44] |
| Termotoga maritima | 35,000 | 200 | 40 | [43] |
| Haloferax volcanii | 11 | 370 | 520 | [44] |
| Giardia lamblia | 31 | 18 | 14 | [53] |
| Plasmodium falciparum | 8 | 19 | 6 | [54] |
| Trypanosoma cruzy | 62 | 77 | 16 | [52] |
| Aspergillus niger | NR | 153 | 26 | [55] |
| Aspergillus oryzae | 1000 | 109 | 6 | [56] |
| Brugia malayi | 40 | 245 | 14 | [49] |
| Dog liver | NR | 122 | 10 | [57] |
| Buffalo liver | NR | NR | 59 | [48] |
| Camel liver | NR | 81 | 81 | [47] |
| Homo sapiens | 230 | 38 | 7 | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Ramírez, P.; Hernández-Ochoa, B.; Ortega-Cuellar, D.; González-Valdez, A.; Martínez-Rosas, V.; Morales-Luna, L.; Arreguin-Espinosa, R.; Castillo-Rodríguez, R.A.; Canseco-Ávila, L.M.; Cárdenas-Rodríguez, N.; et al. Biochemical and Kinetic Characterization of the Glucose-6-Phosphate Dehydrogenase from Helicobacter pylori Strain 29CaP. Microorganisms 2022, 10, 1359. https://doi.org/10.3390/microorganisms10071359
Ortiz-Ramírez P, Hernández-Ochoa B, Ortega-Cuellar D, González-Valdez A, Martínez-Rosas V, Morales-Luna L, Arreguin-Espinosa R, Castillo-Rodríguez RA, Canseco-Ávila LM, Cárdenas-Rodríguez N, et al. Biochemical and Kinetic Characterization of the Glucose-6-Phosphate Dehydrogenase from Helicobacter pylori Strain 29CaP. Microorganisms. 2022; 10(7):1359. https://doi.org/10.3390/microorganisms10071359
Chicago/Turabian StyleOrtiz-Ramírez, Paulina, Beatriz Hernández-Ochoa, Daniel Ortega-Cuellar, Abigail González-Valdez, Víctor Martínez-Rosas, Laura Morales-Luna, Roberto Arreguin-Espinosa, Rosa Angélica Castillo-Rodríguez, Luis Miguel Canseco-Ávila, Noemi Cárdenas-Rodríguez, and et al. 2022. "Biochemical and Kinetic Characterization of the Glucose-6-Phosphate Dehydrogenase from Helicobacter pylori Strain 29CaP" Microorganisms 10, no. 7: 1359. https://doi.org/10.3390/microorganisms10071359