
Emergence of Novel Chlamydia trachomatis Sequence Types among Chlamydia Patients in the Republic of Belarus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
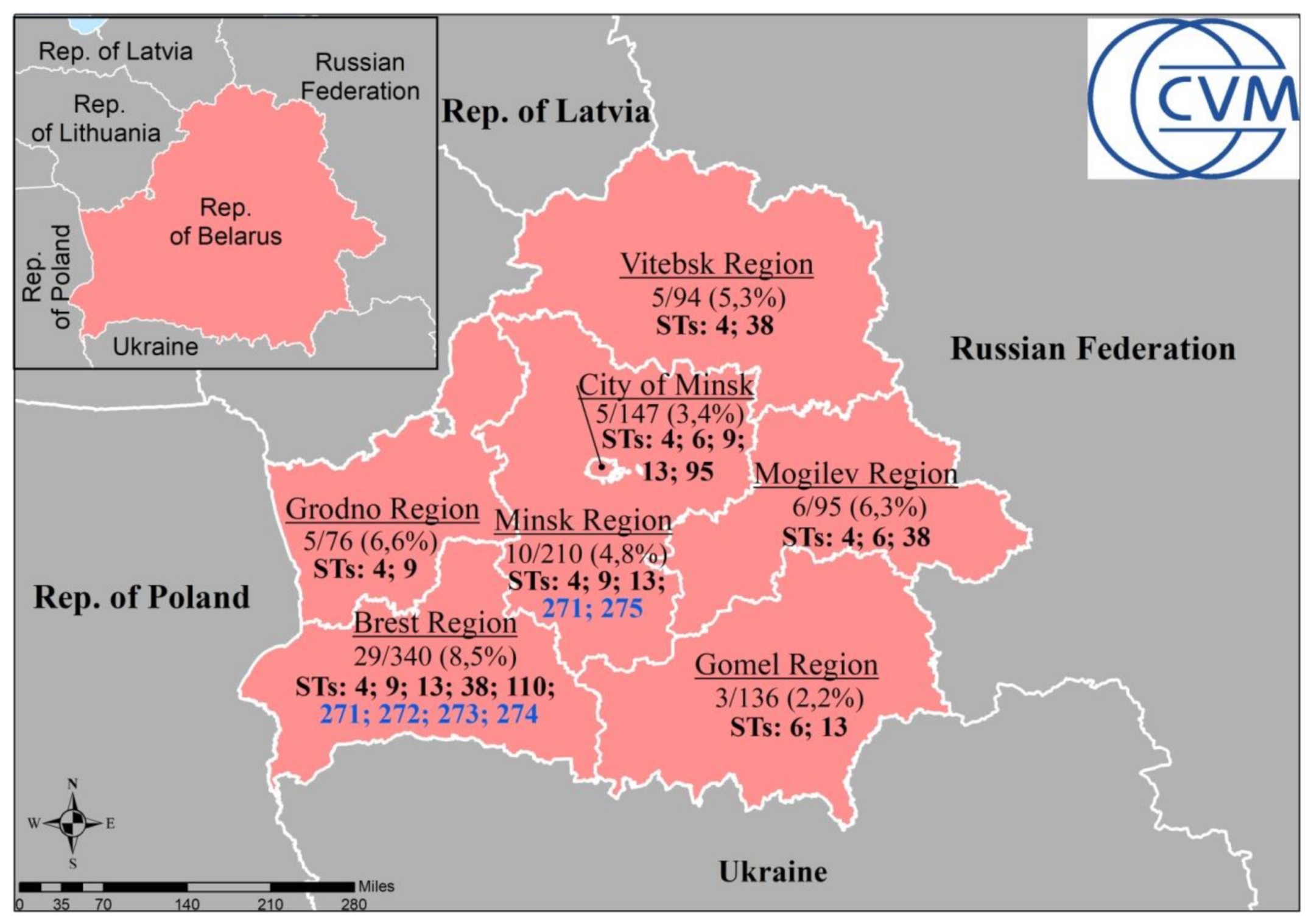
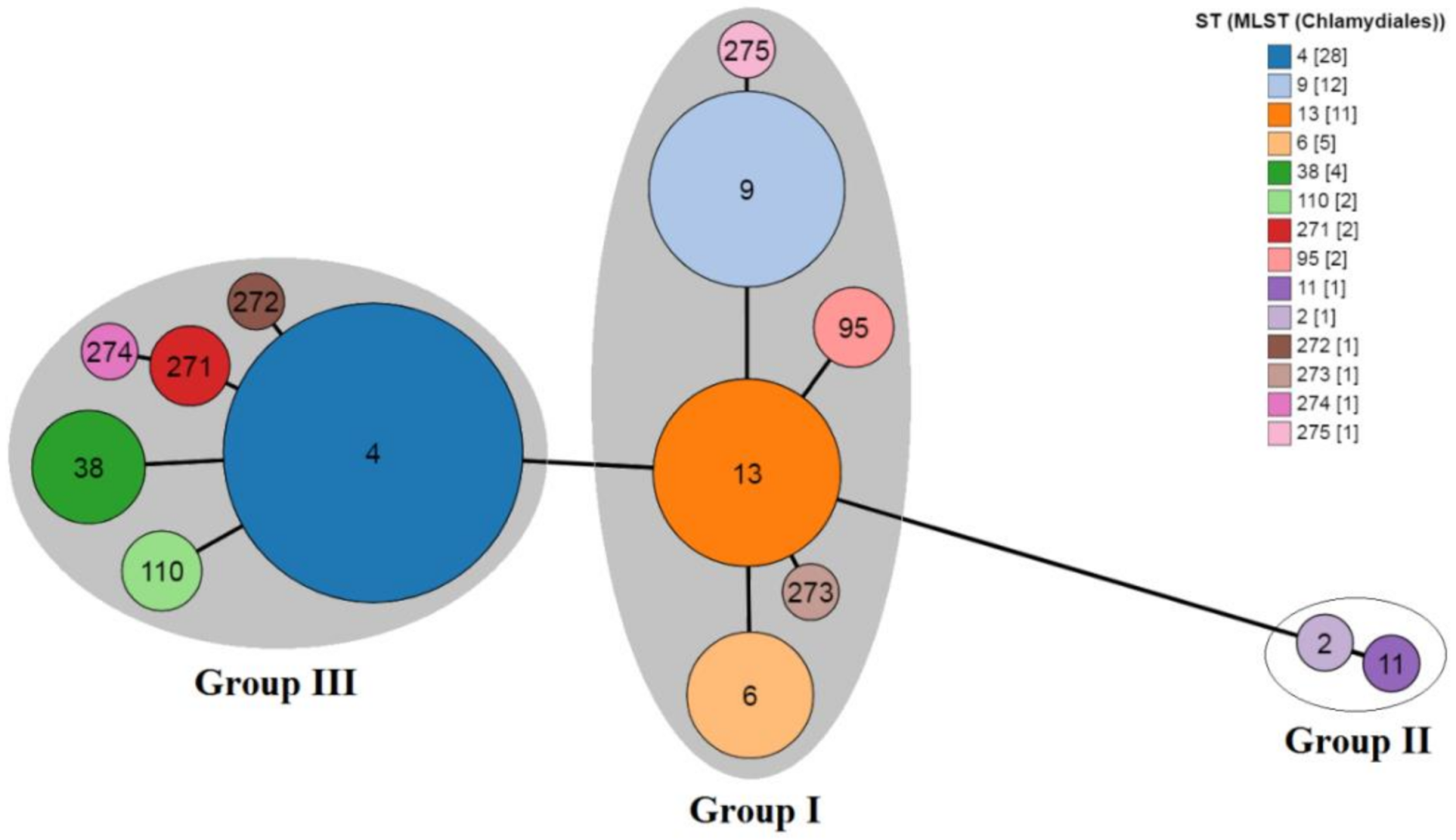
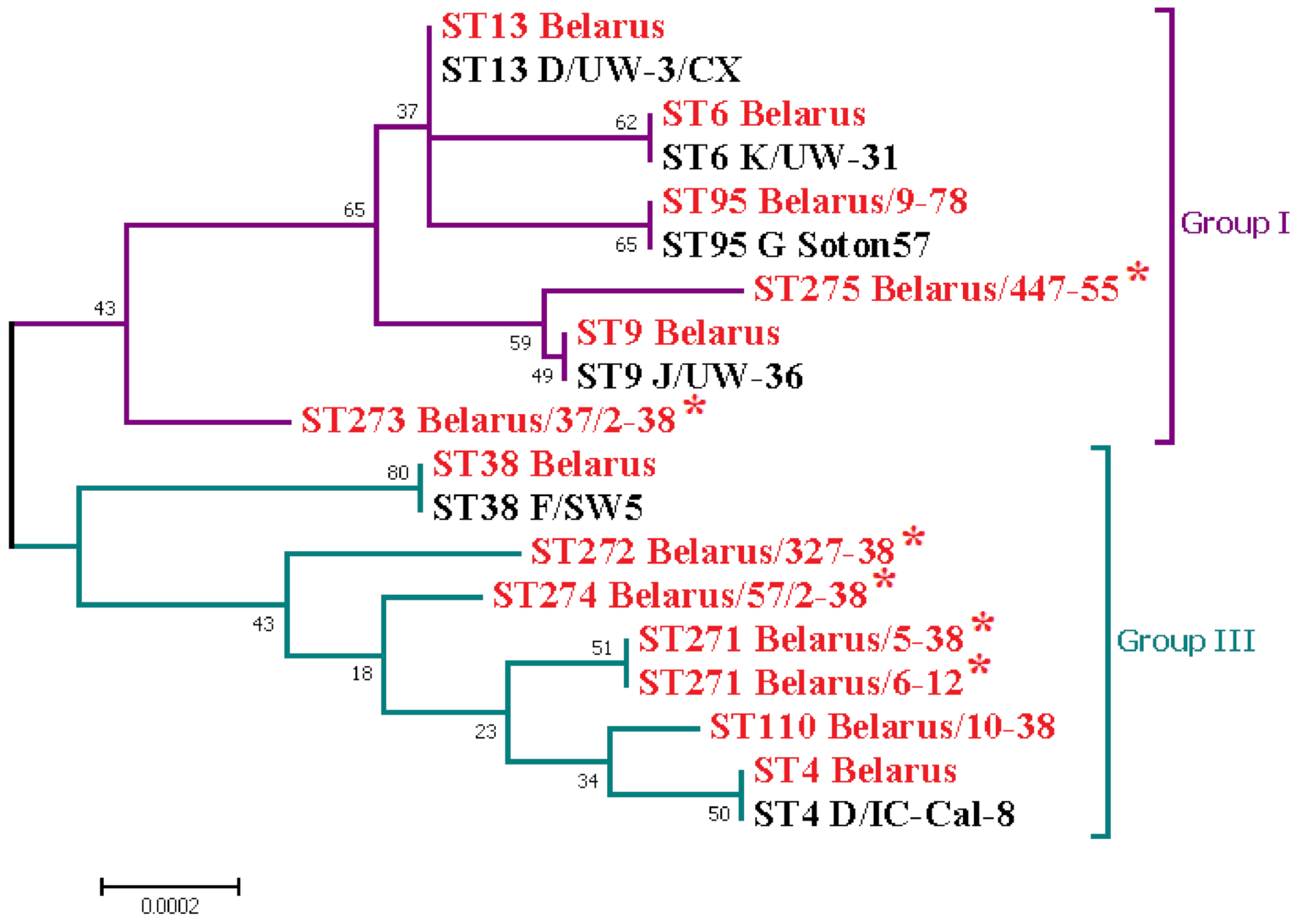
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowley, J.; Vander Hoorn, S.; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.K.; Searle, K.M.; Yang, W.; Rapheal, E.; Wang, C.; Zhao, P.; Yang, L.; Huang, S.; Yang, B. Spatiotemporal analysis of 11 years of Chlamydia trachomatis data from southern China. Lancet Reg. Health West. Pac. 2021, 11, 100143. [Google Scholar] [CrossRef] [PubMed]
- Pannekoek, Y.; Morelli, G.; Kusecek, B.; Morré, A.S.; Ossewaarde, J.M.; Langerak, A.A.; van der Ende, A. Multi locus sequence typing of Chlamydiales: Clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feodorova, V.A.; Konnova, S.; Saltykov, Y.; Zaitsev, S.; Subbotina, I.A.; Polyanina, T.I.; Ulyanov, S.S.; Lamers, S.L.; Gaydos, C.A.; Quinn, T.C.; et al. Urogenital Chlamydia trachomatis multilocus sequence types and genovar distribution in chlamydia infected patients in a multi-ethnic region of Saratov, Russia. PLoS ONE 2018, 13, e0195386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Health Sector Strategy on Sexually Transmitted Infections 2016–2021. Towards Ending STIs; World Health Organization: Geneva, Switzerland, 2016.
- Patiño, L.H.; Camargo, M.; Muñoz, M.; Ríos-Chaparro, D.I.; Patarroyo, M.A.; Ramírez, J.D. Unveiling the Multilocus Sequence Typing (MLST) Schemes and Core Genome Phylogenies for Genotyping Chlamydia trachomatis. Front. Microbiol. 2018, 22, 1854. [Google Scholar] [CrossRef]
- Floridia-Yapur, T.; Rusman, F.; Diosque, P.; Tomasini, N. Genome data vs MLST for exploring intraspecific evolutionary history in bacteria: Much is not always better. Infect. Genet. Evol. 2021, 93, 104990. [Google Scholar] [CrossRef] [PubMed]
- Klint, M.; Fuxelius, H.-H.; Goldkuhl, R.R.; Skarin, H.; Rutemark, C.; Andersson, S.G.E.; Persson, K.; Herrmann, B. High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J. Clin. Microbiol. 2007, 45, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.N.; Herrmann, B.; Møller, J.K. Typing Chlamydia trachomatis: From egg yolk to nanotechnology. FEMS Immunol. Med. Microbiol. 2009, 55, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Jordan, I.K.; Rishishwar, L. stringMLST: A fast k-mer based tool for multilocus sequence typing. Bioinformatic. 2017, 33, 119–121. [Google Scholar] [CrossRef]
- Ikryannikova, L.N.; Shkarupeta, M.M.; Shitikov, E.A.; Il’ina, E.N.; Govorun, V.M. Comparative evaluation of new typing schemes for urogenital Chlamydia trachomatis isolates. FEMS Immunol. Med. Microbiol. 2010, 59, 188–196. [Google Scholar] [CrossRef]
- Ulianova, O.; Ulyanov, S.; Zaytsev, S.; Saltykov, Y.; Ulyanov, A.; Feodorova, V. Could LASCA-imaging of GB-speckles be applied for a high discrimination and typing of pathogenic bacteria? PLoS ONE 2021, 16, e0245657. [Google Scholar] [CrossRef] [PubMed]
- Pilo, S.; Valenci, G.Z.; Rubinstein, M.; Pichadze, L.; Scharf, Y.; Dveyrin, Z.; Rorman, E.; Nissan, I. High-resolution multilocus sequence typing for Chlamydia trachomatis: Improved results for clinical samples with low amounts of C. trachomatis DNA. BMC Microbiol. 2021, 18, 28. [Google Scholar] [CrossRef] [PubMed]
- Christerson, L.; Ruettger, A.; Gravningen, K.; Ehricht, R.; Sachse, K.; Herrmann, B. High-resolution genotyping of Chlamydia trachomatis by use of a novel multilocus typing DNA microarray. J. Clin. Microbiol. 2011, 49, 2838–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksson, J.; Vaulet, L.G.; Christerson, L.; Ruettger, A.; Sachse, K.; Entrocassi, C.; Castro, É.; Fermepin, M.R.; Herrmann, B. Comparison of multilocus sequence typing and multilocus typing microarray of Chlamydia trachomatis strains from Argentina and Chile. J. Microbiol. Methods. 2016, 127, 214–218. [Google Scholar] [CrossRef]
- Bom, R.J.M.; Christerson, L.; van der Loeff, M.F.S.; Coutinho, R.A.; Herrmann, B.; Bruisten, S.M. Evaluation of high-resolution typing methods for Chlamydia trachomatis in samples from heterosexual couples. J. Clin. Microbiol. 2011, 49, 2844–2853. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, B.; Isaksson, J.; Ryberg, M.; Tångrot, J.; Saleh, I.; Versteeg, B.; Gravningen, K.; Bruisten, S. Global Multilocus Sequence Type Analysis of Chlamydia trachomatis Strains from 16 Countries. J. Clin. Microbiol. 2015, 53, 2172–2179. [Google Scholar] [CrossRef] [Green Version]
- Dean, D.; Bruno, W.J.; Wan, R.; Gomes, J.P.; Devignot, S.; Mehari, T.; De Vries, H.J.; Morré, S.A.; Myers, G.; Read, T.D.; et al. Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg. Infect. Dis. 2009, 15, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, B.; Törner, A.; Low, N.; Klint, M.; Nilsson, A.; Veličko, I.; Söderblom, T.; Blaxhult, A. Emergence and spread of Chlamydia trachomatis variant, Sweden. Emerg. Infect. Dis. 2008, 14, 1462–1465. [Google Scholar] [CrossRef] [Green Version]
- Jurstrand, M.; Christerson, L.; Klint, M.; Fredlund, H.; Unemo, M.; Herrmann, B. Characterisation of Chlamydia trachomatis by ompA sequencing and multilocus sequence typing in a Swedish county before and after identification of the new variant. Sex. Transm. Infect. 2010, 86, 56–60. [Google Scholar] [CrossRef]
- Gravningen, K.; Christerson, L.; Furberg, A.-S.; Simonsen, G.S.; Odman, K.; Stahlsten, A.; Herrmann, B. Multilocus sequence typing of genital Chlamydia trachomatis in Norway reveals multiple new sequence types and a large genetic diversity. PLoS ONE 2012, 7, e34452. [Google Scholar] [CrossRef]
- Christerson, L.; Bom, R.J.M.; Bruisten, S.M.; Yass, R.; Hardick, J.; Bratt, G.; Gaydos, C.A.; Morré, S.A.; Herrmann, B. Chlamydia trachomatis strains show specific clustering for men who have sex with men compared to heterosexual populations in Sweden, the Netherlands, and the United States. J. Clin. Microbiol. 2012, 50, 3548–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bom, R.J.M.; Van Der Helm, J.J.; Van Der Loeff, M.F.S.; Van Rooijen, M.S.; Heijman, T.; Matser, A.; De Vries, H.J.C.; Bruisten, S.M. Distinct transmission networks of Chlamydia trachomatis in men who have sex with men and heterosexual adults in Amsterdam, The Netherlands. PLoS ONE 2013, 8, e53869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christerson, L.; De Vries, H.J.; De Barbeyrac, B.; Gaydos, C.A.; Henrich, B.; Hoffmann, S.; Schachter, J.; Thorvaldsen, J.; Vall-Mayans, M.; Klint, M.; et al. Typing of lymphogranuloma venereum Chlamydia trachomatis strains. Emerg. Infect. Dis. 2010, 16, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, J.; Carlsson, O.; Airell, Å.; Strömdahl, S.; Bratt, G.; Herrmann, B. Lymphogranuloma venereum rates increased and Chlamydia trachomatis genotypes changed among men who have sex with men in Sweden 2004–2016. J. Med. Microbiol. 2017, 66, 1684–1687. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, B.; van Rooijen, M.S.; Schim van der Loeff, M.F.; de Vries, H.J.C.; Bruisten, S.M. No indication for tissue tropism in urogenital and anorectal Chlamydia trachomatis infections using high-resolution multilocus sequence typing. BMC Infect. Dis. 2014, 14, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding-Esch, E.M.; Holland, M.J.; Schémann, J.-F.; Sillah, A.; Sarr, B.; Christerson, L.; Pickering, H.; Molina-Gonzalez, S.; Sarr, I.; Andreasen, A.A.; et al. Impact of a single round of mass drug administration with azithromycin on active trachoma and ocular Chlamydia trachomatis prevalence and circulating strains in The Gambia and Senegal. Parasit. Vectors 2019, 12, 497. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, L.; Isaksson, J.; Zapico, M.; Cilla, G.; Herrmann, B. Chlamydia trachomatis genotypes A and B from urogenital specimens of patients in Spain: Molecular characterization. Clin. Microbiol. Infect. 2018, 24, 910.e5–910.e8. [Google Scholar] [CrossRef] [Green Version]
- Feodorova, V.A.; Zaitsev, S.S.; Saltykov, Y.V.; Ulyanov, S.S.; Motin, V.L. Multi-locus sequence analysis reveals a novel sequence type of Chlamydia trachomatis in Saratov Region, Russia. New Microbes New Infect. 2019, 31, 100584. [Google Scholar] [CrossRef]
- Feodorova, V.A.; Zaitsev, S.S.; Saltykov, Y.V.; Sultanakhmedov, E.S.; Bakulev, A.L.; Ulyanov, S.S.; Motin, V.L. An Asymptomatic Patient with Fatal Infertility Carried a Swedish Strain of Chlamydia trachomatis with Additional Deletion in The Plasmid orf1 that Belonged to A Different MLST Sequence Type. Microorganisms 2019, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- The Ministry of Health of the Republic of Belarus. Public Health in the Republic of Belarus: An Official Statistics Collection, 2018; The Ministry of Health of the Republic of Belarus: Minsk, Belarus, 2019; p. 261.
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, M.; Sung, S.; Takov, V. Chlamydia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022; p. NBK537286. [Google Scholar] [PubMed]
- Christerson, L.; de Vries, H.J.C.; Klint, M.; Herrmann, B.; Morré, S.A. Multilocus sequence typing of urogenital Chlamydia trachomatis from patients with different degrees of clinical symptoms. Sex. Transm. Dis. 2011, 38, 490–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, B.; Himschoot, M.; van den Broek, I.V.F.; Bom Reinier, J.M.; Speksnijder, A.G.C.L.; van der Loeff, M.F.S.; Bruisten, S.M. Urogenital Chlamydia trachomatis strain types, defined by high-resolution multilocus sequence typing, in relation to ethnicity and urogenital symptoms among a young screening population in Amsterdam, The Netherlands. Sex. Transm. Infect. 2015, 91, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; Clarke, I.N.; Seth-Smith, H.M.; Solomon, A.W.; Cutcliffe, L.T.; Marsh, P.; Skilton, R.J.; Holland, M.J.; Mabey, D.; Peeling, R.W.; et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet. 2012, 44, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Harris, S.R.; Seth-Smith, H.M.B.; Parmar, S.; Andersson, P.; Giffard, P.M.; Schachter, J.; Moncada, J.; Ellison, L.; Vaulet, M.L.G.; et al. Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 2017, 27, 1220–1229. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feodorova, V.A.; Saltykov, Y.V.; Kolosova, A.A.; Rubanik, L.V.; Poleshchuk, N.N.; Motin, V.L. Emergence of Novel Chlamydia trachomatis Sequence Types among Chlamydia Patients in the Republic of Belarus. Microorganisms 2022, 10, 478. https://doi.org/10.3390/microorganisms10020478
Feodorova VA, Saltykov YV, Kolosova AA, Rubanik LV, Poleshchuk NN, Motin VL. Emergence of Novel Chlamydia trachomatis Sequence Types among Chlamydia Patients in the Republic of Belarus. Microorganisms. 2022; 10(2):478. https://doi.org/10.3390/microorganisms10020478
Chicago/Turabian StyleFeodorova, Valentina A., Yury V. Saltykov, Anna A. Kolosova, Liudmila V. Rubanik, Nikolay N. Poleshchuk, and Vladimir L. Motin. 2022. "Emergence of Novel Chlamydia trachomatis Sequence Types among Chlamydia Patients in the Republic of Belarus" Microorganisms 10, no. 2: 478. https://doi.org/10.3390/microorganisms10020478