
Bermudagrass Cultivars with Different Tolerance to Nematode Damage Are Characterized by Distinct Fungal but Similar Bacterial and Archaeal Microbiomes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bermudagrass and Soil Sampling
2.2. Sample Preparation
2.3. DNA Extraction, 16S rRNA Gene V4 and ITS2 Amplicon PCR and Sequencing
2.4. Isolation and Identification of Endophytic Fungi
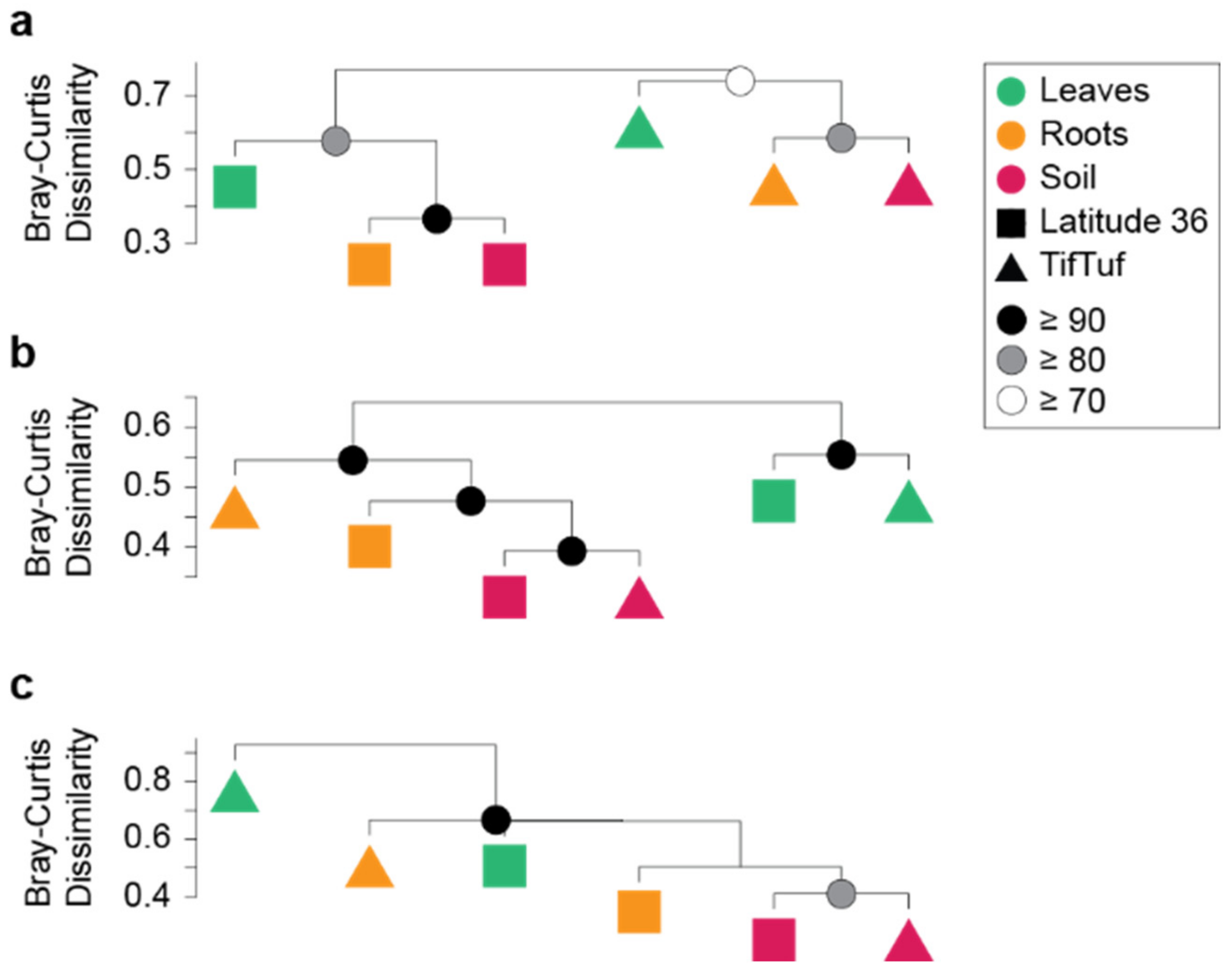
2.5. Hierarchical Clustering
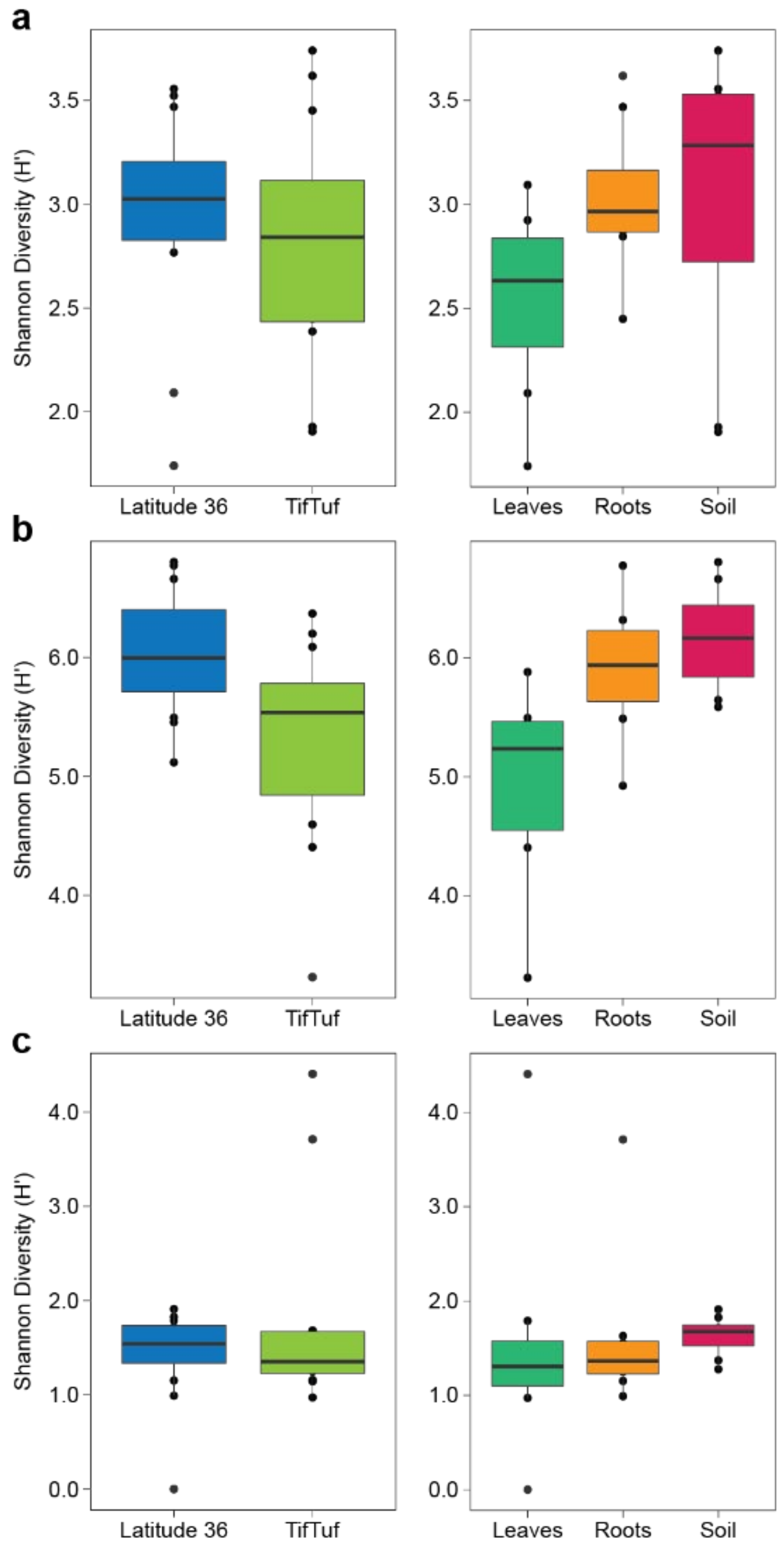
2.6. Microbial Diversity and Indicator Species Analyses
2.7. Data Availability and Deposition
3. Results and Discussion
3.1. A Unique System for the Study of Turfgrass Microbiomes and Host Functions
3.2. General Patterns in Microbiome Structure between Cultivars and Microhabitats
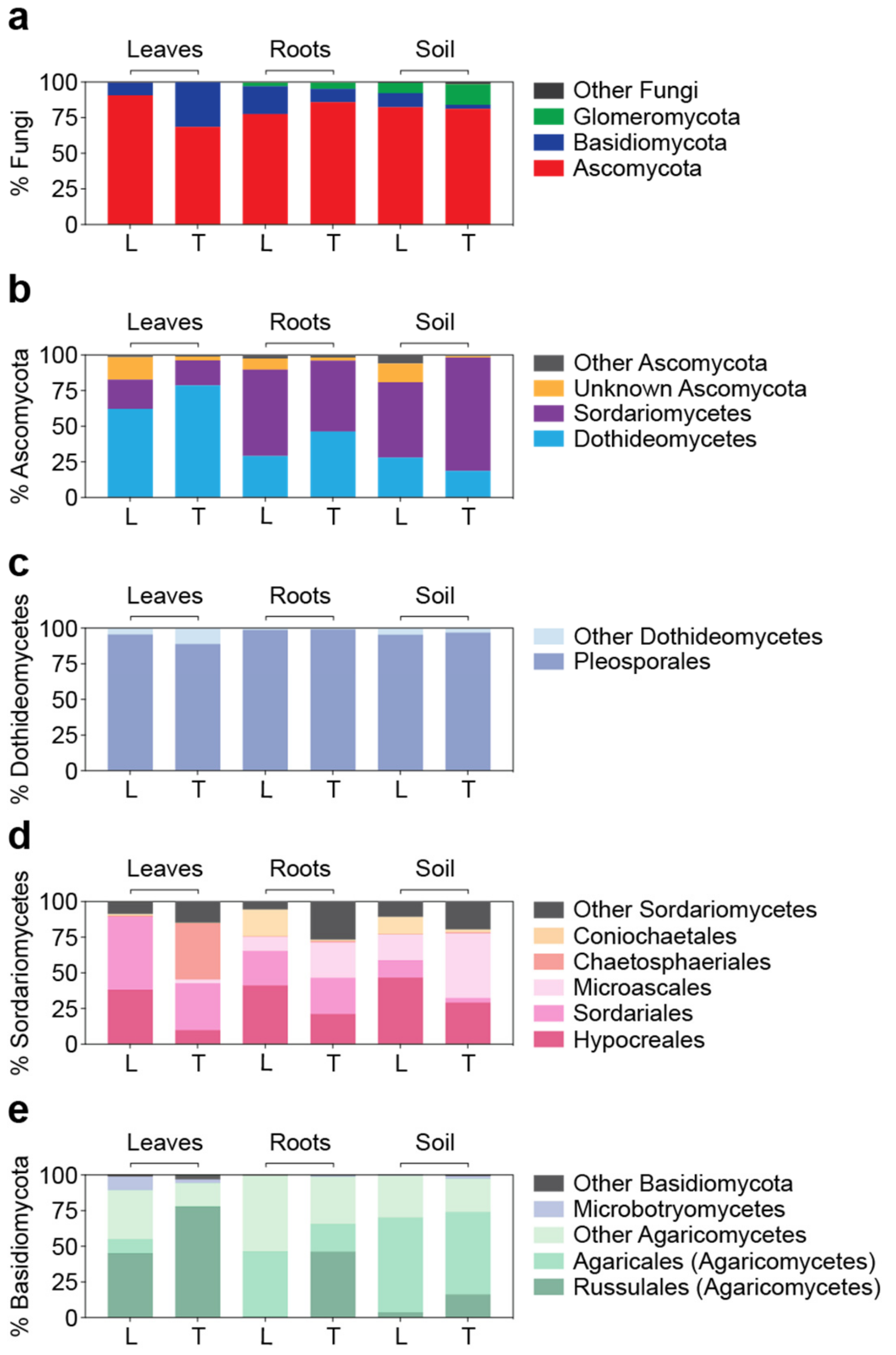
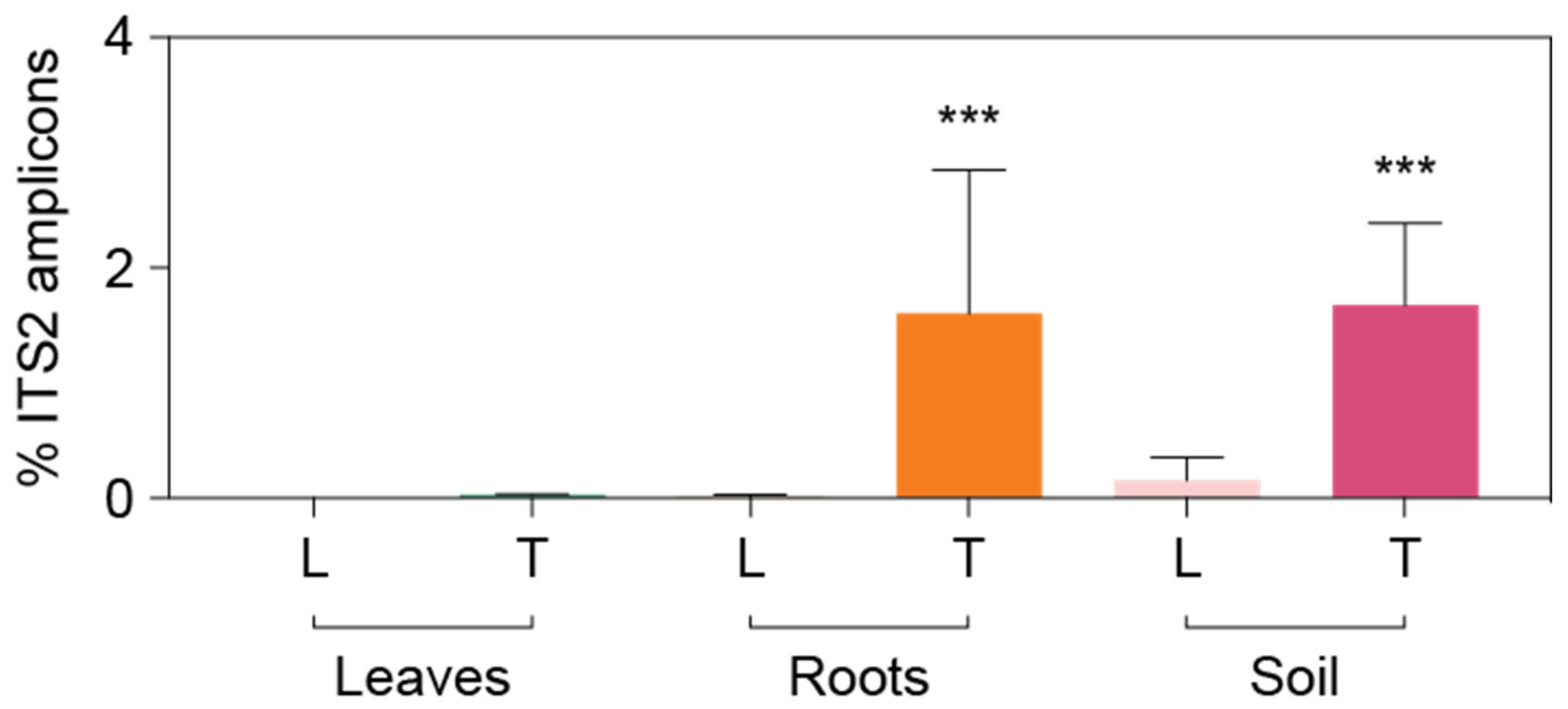
3.3. Detailed Microbial Community Structure in Bermudagrasses Exhibiting Different Susceptibility to Nematode Infection: Fungal Microbiomes
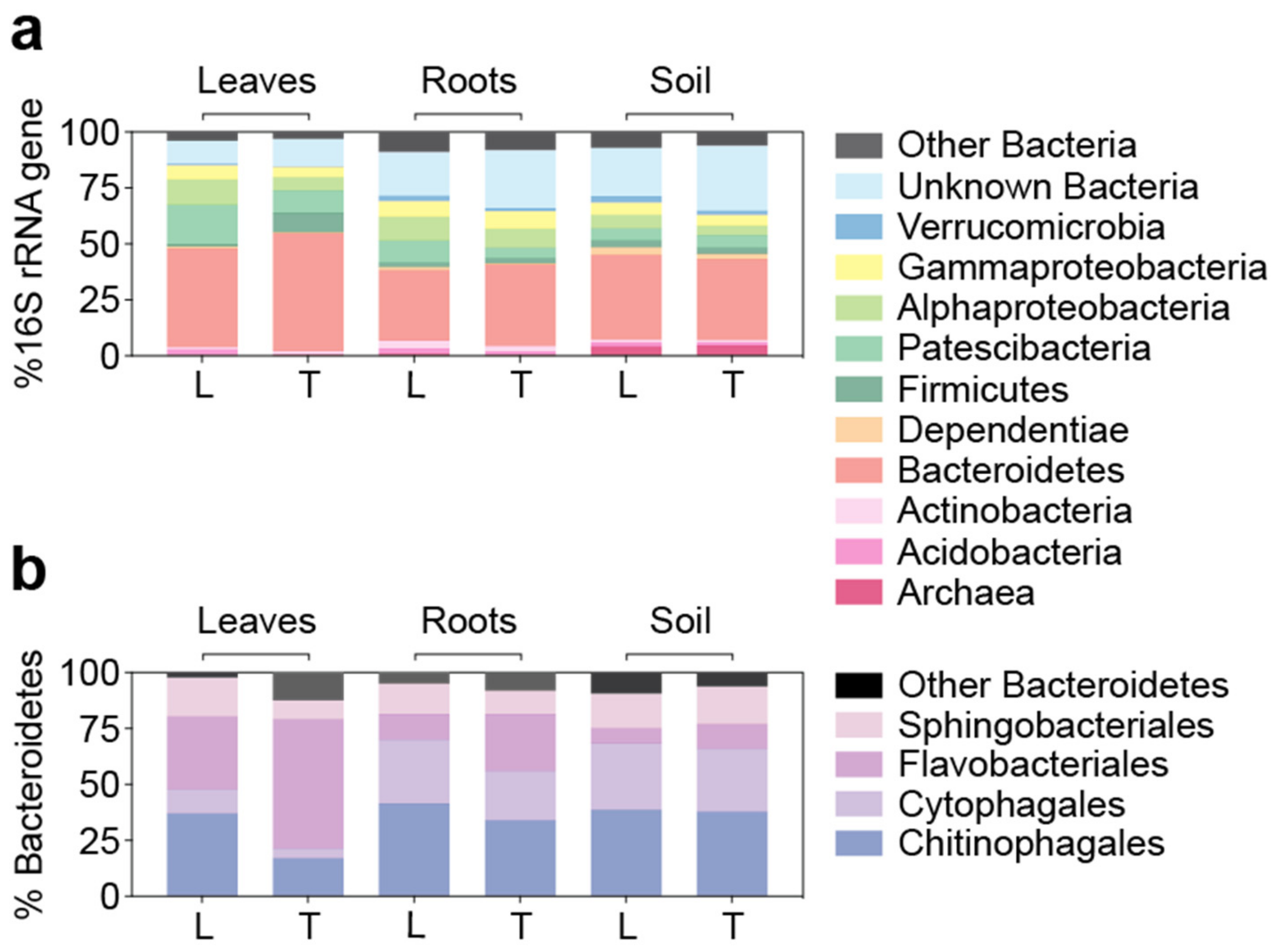
3.4. Similar Bacterial Microbiomes in Latitude 36 and TifTuf
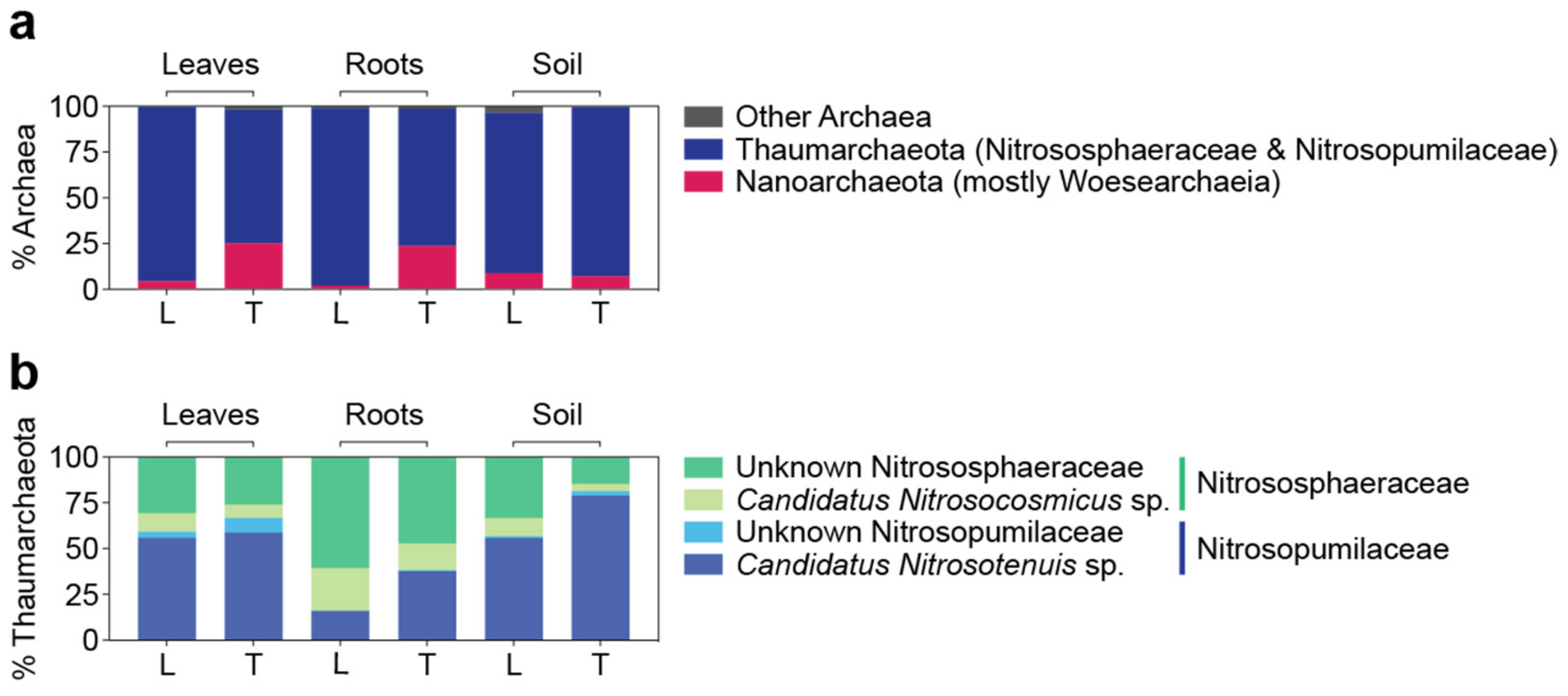
3.5. Archaeal Microbiomes in Bermudagrasses: Potential Link to Nitrification
3.6. Culturable Endophytic Microbes Isolated from the Bermudagrass Cultivars
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haase, D.; Larondelle, N.; Andersson, E.; Artmann, M.; Borgström, S.; Breuste, J.; Gomez-Baggethun, E.; Gren, Å.; Hamstead, Z.; Hansen, R. A quantitative review of urban ecosystem service assessments: Concepts, models, and implementation. Ambio 2014, 43, 413–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Follett, R. Carbon dynamics and sequestration in urban turfgrass ecosystems. In Carbon Sequestration in Urban Ecosystems; Springer: Dordrecht, Germany, 2012; pp. 161–172. [Google Scholar]
- Ignatieva, M.; Hedblom, M. An alternative urban green carpet. Science 2018, 362, 148–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, J.B.; Green, R.L. The role of turfgrasses in environmental protection and their benefits to humans. J. Environ. Qual. 1994, 23, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.A. Ecosystem services from turfgrass landscapes. Urban For. Urban Green. 2017, 26, 151–157. [Google Scholar] [CrossRef]
- Qian, Y.; Bandaranayake, W.; Parton, W.; Mecham, B.; Harivandi, M.; Mosier, A. Long-term effects of clipping and nitrogen management in turfgrass on soil organic carbon and nitrogen dynamics: The CENTURY model simulation. J. Environ. Qual. 2003, 32, 1694–1700. [Google Scholar] [CrossRef]
- Milesi, C.; Running, S.W.; Elvidge, C.D.; Dietz, J.B.; Tuttle, B.T.; Nemani, R.R. Mapping and modeling the biogeochemical cycling of turf grasses in the United States. Environ. Manag. 2005, 36, 426–438. [Google Scholar] [CrossRef]
- Singh, B.K.; Trivedi, P.; Egidi, E.; Macdonald, C.A.; Delgado-Baquerizo, M. Crop microbiome and sustainable agriculture. Nat. Rev. Microbiol. 2020, 18, 601–602. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Crow, W.T. Turfgrass nematicide and bionematicide research in Florida. Outlooks Pest Manag. 2014, 25, 222–225. [Google Scholar] [CrossRef]
- Vandenbossche, B.; Viaene, N.; Sutter, N.; Maes, M.; Karssen, G.; Bert, W. Diversity and incidence of plant-parasitic nematodes in Belgian turf grass. Nematology 2011, 13, 245–256. [Google Scholar] [CrossRef]
- Walker, N.; Goad, C.; Zhang, H.; Martin, D. Factors associated with populations of plant-parasitic nematodes in bentgrass putting greens in Oklahoma. Plant Dis. 2002, 86, 764–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, W.; Luc, J.E.; Crow, W.T.; Kenworthy, K.E.; Giblin-Davis, R.M.; McSorley, R.; Kruse, J.K. Bermudagrass cultivar responses to sting nematodes. Crop Sci. 2011, 51, 2199–2203. [Google Scholar] [CrossRef]
- Leuchtmann, A.; Bacon, C.W.; Schardl, C.L.; White, J.F., Jr.; Tadych, M. Nomenclatural realignment of Neotyphodium species with genus Epichloë. Mycologia 2014, 106, 202–215. [Google Scholar] [CrossRef]
- Kimmons, C.; Gwinn, K.; Bernard, E. Nematode reproduction on endophyte-infected and endophyte-free tall fescue. Plant Dis. 1990, 74, 757–761. [Google Scholar] [CrossRef]
- Elmi, A.; West, C.; Robbins, R.; Kirkpatrick, T. Endophyte effects on reproduction of a root-knot nematode (Meloidogyne marylandi) and osmotic adjustment in tall fescue. Grass Forage Sci. 2000, 55, 166–172. [Google Scholar] [CrossRef]
- Jiang, X.; Xiang, M.; Liu, X. Nematode-trapping fungi. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Lian, L.; Tian, B.; Xiong, R.; Zhu, M.; Xu, J.; Zhang, K. Proteases from Bacillus: A new insight into the mechanism of action for rhizobacterial suppression of nematode populations. Lett. Appl. Microbiol. 2007, 45, 262–269. [Google Scholar] [CrossRef]
- Oka, Y.; Chet, I.; Spiegel, Y. Control of the rootknot nematode Meloidogyne javanica by Bacillus cereus. Biocontrol Sci. Technol. 1993, 3, 115–126. [Google Scholar] [CrossRef]
- Siddiqui, Z.A.; Qureshi, A.; Akhtar, M. Biocontrol of root-knot nematode Meloidogyne incognita by Pseudomonas and Bacillus isolates on Pisum sativum. Arch. Phytopathol. Plant Prot. 2009, 42, 1154–1164. [Google Scholar] [CrossRef]
- Liang, L.-M.; Zou, C.-G.; Xu, J.; Zhang, K.-Q. Signal pathways involved in microbe–nematode interactions provide new insights into the biocontrol of plant-parasitic nematodes. Philos. Trans. R. Soc. B 2019, 374, 20180317. [Google Scholar] [CrossRef]
- Crow, W. Nematode management for golf courses in Florida. EDIS 2020, ENY-008, 1–8. [Google Scholar] [CrossRef]
- Crow, W.T.; Luc, J.E.; Sekora, N.S.; Pang, W. Interaction between Belonolaimus longicaudatus and Helicotylenchus pseudorobustus on bermudagrass and seashore paspalum hosts. J. Nematol. 2013, 45, 17. [Google Scholar] [PubMed]
- McPherson, M.R.; Wang, P.; Marsh, E.L.; Mitchell, R.B.; Schachtman, D.P. Isolation and analysis of microbial communities in soil, rhizosphere, and roots in perennial grass experiments. J. Vis. Exp. 2018, 137, e57932. [Google Scholar] [CrossRef] [Green Version]
- Richter-Heitmann, T.; Eickhorst, T.; Knauth, S.; Friedrich, M.W.; Schmidt, H. Evaluation of strategies to separate root-associated microbial communities: A crucial choice in rhizobiome research. Front. Microbiol. 2016, 7, 773. [Google Scholar] [CrossRef] [PubMed]
- Mylavarapu, R. UF/IFAS extension soil testing laboratory (ESTL) analytical procedures and training manual. EDIS 2009, 2009, 14. [Google Scholar] [CrossRef]
- Crow, W.T.; Habteweld, A.; Bean, T. Mist chamber extraction for improved diagnosis of Meloidogyne spp. from golf course bermudagrass. J. Nematol. 2020, 52, e2020-96. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 January 2022).
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. Available online: https://github.com/najoshi/sickle (accessed on 5 January 2022).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, K.K.; Peterson, S.W.; Jong, S.-C. Preservation and distribution of fungal cultures. In Biodiversity of Fungi: Inventory and Monitoring Methods; Elsevier Academic Press: Amsterdam, The Netherlands, 2004; pp. 37–47. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Vilgalys, R.; Hester, M. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 1990, 172, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Roberts, D.W.; Package ‘labdsv’. Ordination and Multivariate. Available online: https://cran.r-project.org/web/packages/labdsv/index.html (accessed on 5 January 2022).
- Stingl, U.; Choi, C.J.; Dhillon, B.; Schiavon, M. The Lack of Knowledge on the Microbiome of Golf Turfgrasses Impedes the Development of Successful Microbial Products. Agronomy 2022, 12, 71. [Google Scholar] [CrossRef]
- Xia, Q.; Rufty, T.; Shi, W. Predominant microbial colonizers in the root endosphere and rhizosphere of turfgrass systems: Pseudomonas veronii, Janthinobacterium lividum, and Pseudogymnoascus spp. Front. Microbiol. 2021, 12, 643904. [Google Scholar] [CrossRef]
- Crouch, J.A.; Carter, Z.; Ismaiel, A.; Roberts, J.A. The US National Mall microbiome: A census of rhizosphere bacteria inhabiting landscape turf. Crop Sci. 2017, 57, S341–S348. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Luo, Y.; Sun, P.; Gao, J.; Zhao, D.; Yang, P.; Hu, T. Effects of shade stress on turfgrasses morphophysiology and rhizosphere soil bacterial communities. BMC Plant Biol. 2020, 20, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, E.; Larsson, K.-H. Phylogenetic relationships of russuloid basidiomycetes with emphasis on aphyllophoralean taxa. Mycologia 2003, 95, 1037–1065. [Google Scholar] [CrossRef] [PubMed]
- Mc Cargo, P.D.; Iannone, L.J.; Soria, M.; Novas, M.V. Diversity of foliar endophytes in a dioecious wild grass and their interaction with the systemic Epichloë. Fungal Ecol. 2020, 47, 100945. [Google Scholar] [CrossRef]
- Rudgers, J.A.; Fox, S.; Porras-Alfaro, A.; Herrera, J.; Reazin, C.; Kent, D.R.; Souza, L.; Chung, Y.A.; Jumpponen, A. Biogeography of root-associated fungi in foundation grasses of North American plains. J. Biogeogr. 2022, 49, 22–37. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Herrera, J.; Sinsabaugh, R.L.; Odenbach, K.J.; Lowrey, T.; Natvig, D.O. Novel root fungal consortium associated with a dominant desert grass. Appl. Environ. Microbiol. 2008, 74, 2805–2813. [Google Scholar] [CrossRef] [Green Version]
- Mouhamadou, B.; Puissant, J.; Personeni, E.; Desclos-Theveniau, M.; Kastl, E.; Schloter, M.; Zinger, L.; Roy, J.; Geremia, R.; Lavorel, S. Effects of two grass species on the composition of soil fungal communities. Biol. Fertil. Soils 2013, 49, 1131–1139. [Google Scholar] [CrossRef]
- Chen, L.; Saixi, Y.; Yi, R.; Baoyin, T. Characterization of soil microbes associated with a grazing-tolerant grass species, Stipa breviflora, in the Inner Mongolian desert steppe. Ecol. Evol. 2020, 10, 10607–10618. [Google Scholar] [CrossRef]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef]
- Tourna, M.; Stieglmeier, M.; Spang, A.; Könneke, M.; Schintlmeister, A.; Urich, T.; Engel, M.; Schloter, M.; Wagner, M.; Richter, A.; et al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. USA 2011, 108, 8420–8425. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.B.; De La Torre, J.; Klotz, M.; Urakawa, H.; Pinel, N.; Arp, D.; Brochier-Armanet, C.; Chain, P.; Chan, P.; Gollabgir, A. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8818–8823. [Google Scholar] [CrossRef] [Green Version]
- Stahl, D.A.; de la Torre, J.R. Physiology and diversity of ammonia-oxidizing archaea. Annu. Rev. Microbiol. 2012, 66, 83–101. [Google Scholar] [CrossRef]
- Schiavon, M.; Shaddox, T.W.; Williams, K.E.; Gallo, S.; Boeri, P.A.; Unruh, J.B.; Kruse, J.; Kenworthy, K. Nitrogen requirements for deficit-irrigated bermudagrass (Cynodon spp.) fairways in South Florida. J. Agron. Crop Sci. online ahead of print. 2021. [Google Scholar] [CrossRef]
- Schardl, C.L.; Leuchtmann, A.; Spiering, M.J. Symbioses of grasses with seedborne fungal endophytes. Annu. Rev. Plant Biol. 2004, 55, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.; DeGraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 2017, 11, 2691–2704. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Proctor, R.; Brown, D. Comparative genomic analyses of secondary metabolite biosynthetic gene clusters in 207 isolates of Fusarium. In Proceedings of the 29th Fungal Genetics Conference, Genetics Society of America, Pacific Grove, CA, USA, 14–19 March 2017. [Google Scholar]
- Jacobs-Venter, A.; Laraba, I.; Geiser, D.M.; Busman, M.; Vaughan, M.M.; Proctor, R.H.; McCormick, S.P.; O’Donnell, K. Molecular systematics of two sister clades, the Fusarium concolor and F. babinda species complexes, and the discovery of a novel microcycle macroconidium–producing species from South Africa. Mycologia 2018, 110, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Ciancio, A. Observations on the nematicidal properties of some mycotoxins. Fundam. Appl. Nematol. 1995, 18, 451–454. [Google Scholar]
- Radhakrishnan, R.; Hashem, A.; Abd_Allah, E.F. Bacillus: A biological tool for crop improvement through bio-molecular changes in adverse environments. Front. Physiol. 2017, 8, 667. [Google Scholar] [CrossRef]
- Choi, T.G.; Maung, C.E.H.; Lee, D.R.; Henry, A.B.; Lee, Y.S.; Kim, K.Y. Role of bacterial antagonists of fungal pathogens, Bacillus thuringiensis KYC and Bacillus velezensis CE 100 in control of root-knot nematode, Meloidogyne incognita and subsequent growth promotion of tomato. Biocontrol Sci. Technol. 2020, 30, 685–700. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant—Parasitic Nematodes. | Latitude 36 | TifTuf |
|---|---|---|
| RKN (Meloidogyne spp.) J2 | 204 ●●● | 20 |
| Sting (Belonolaimus spp.) | 47 ●●● | 14 ●● |
| Lance (Hoplolaimus spp.) | 175 ●● | 95 |
| Stubby (Trichodorus spp.) | 19 | 55 ●● |
| Spiral (Helicotylenchus spp.) | 0 | 12 |
| Lesion (Pratylenchus spp.) | 9 | 0 |
| Ring (Criconemella spp.) | 1 | 22 |
| Sheathoid (Hemicriconemoides spp.) | 2 | 73 |
| Dagger (Xiphinema spp.) | 0 | 11 |
| Pin (Paratylenchus spp.) | 986 | 0 |
| Latitude 36 | TifTuf | |
|---|---|---|
| Al | 179.86 ± 36.96 | 124.28 ± 6.06 * |
| B | 0.60 ± 0.39 | 0.84 ± 0.28 |
| Mn | 42.99 ± 10.20 | 40.44 ± 11.33 |
| Mo | 0.14 ± 0.02 | 0.17 ± 0.02 |
| Ca | 868.92 ± 59.86 | 964.01 ± 33.32 |
| Fe | 146.31 ± 38.09 | 120.29 ± 24.34 |
| K | 11.45 ± 3.85 | 14.16 ± 6.19 |
| Mg | 22.12 ± 1.98 | 23.88 ± 3.26 |
| Na | 11.86 ± 0.97 | 14.16 ± 3.04 |
| NH4+-N | 1.10 ± 0.27 | 1.49 ± 0.94 |
| NO3−-N | 2.72 ± 1.35 | 3.45 ± 1.43 |
| TKN | 666.26 ± 125.27 | 788.12 ± 81.04 |
| P | 69.34 ± 21.21 | 42.00 ± 9.04 * |
| Si | 63.56 ± 1.27 | 50.72 ± 4.72 * |
| Cd | 1.05 ± 0.26 | 0.84 ± 0.01 |
| Cu | 3.76 ± 1.65 | 3.01 ± 0.96 |
| Ni | 1.20 ± 0.12 | 1.01 ± 0.04 |
| Pb | 6.16 ± 2.19 | 4.59 ± 0.51 |
| Zn | 9.18 ± 0.98 | 8.93 ± 2.23 |
| pH | 7.91 ± 0.16 | 7.72 ± 0.05 * |
| ASV | IndVal | p-Value | Significance | Habitat Specificity | Phylum | Class | Order | Family | Genus/Species |
|---|---|---|---|---|---|---|---|---|---|
| Latitude 36 | |||||||||
| ASV58 | 0.632 | 0.0001 | *** |  | Ascomycota | Dothideomycetes | Pleosporales | Halotthiaceae | Sulcosporium sp. |
| ASV276 | 0.602 | 0.0049 | ** |  | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV11 | 0.592 | 0.0004 | *** | Ascomycota | Sordariomycetes | Sordariales | Lasiosphaeriaceae | NA | |
| ASV27 | 0.580 | 0.0001 | *** | | Ascomycota | Dothideomycetes | Pleosporales | Halotthiaceae | Sulcosporium sp. |
| ASV19 | 0.538 | 0.0001 | *** |  | Ascomycota | Dothideomycetes | Pleosporales | Pleosporaceae | NA |
| ASV95 | 0.525 | 0.0017 | ** | | Ascomycota | Sordariomycetes | Hypocreales | Stachybotryaceae | Myrothecium cinctum |
| ASV569 | 0.513 | 0.0133 | * | Ascomycota | Dothideomycetes | Pleosporales | Pleosporaceae | Curvularia sorghina | |
| ASV187 | 0.496 | 0.0135 | * | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | Kamienskia perpusilla |
| ASV398 | 0.494 | 0.0381 | * |  | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | Dominikia sp. |
| ASV42 | 0.492 | 0.0001 | *** | | Ascomycota | Sordariomycetes | Branch06 | NA | NA |
| TifTuf | |||||||||
| ASV258 | 0.609 | 0.0016 | ** |  | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV52 | 0.600 | 0.0016 | ** | | Ascomycota | Sordariomycetes | Hypocreales | Nectriaceae | Fusarium polyphialidicum |
| ASV434 | 0.585 | 0.0064 | ** | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV43 | 0.572 | 0.0018 | ** | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV67 | 0.542 | 0.0001 | *** |  | Ascomycota | Sordariomycetes | Hypocreales | Nectriaceae | Mariannaea sp. |
| ASV145 | 0.541 | 0.0064 | ** | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV272 | 0.495 | 0.0124 | * | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV161 | 0.495 | 0.0150 | * | | Glomeromycota | Glomeromycetes | Glomerales | Glomeraceae | NA |
| ASV25 | 0.473 | 0.0010 | *** |  | Ascomycota | Dothideomycetes | Pleosporales | Didymellaceae | NA |
| ASV49 | 0.426 | 0.0169 | ** | | Ascomycota | Sordariomycetes | Hypocreales | Nectriaceae | Fusarium solani |
 Leaves Leaves  Roots Roots  Soil Soil  Latitude 36 Latitude 36  TifTuf TifTuf | |||||||||
| ASV | IndVal | p-Value | Significance | Habitat Specificity | Phylum | Class | Order | Family | Genus/Species |
|---|---|---|---|---|---|---|---|---|---|
| Latitude 36 | |||||||||
| ASV915 | 0.805 | 0.0001 | *** |  | Patescibacteria | Saccharimonadia | Saccharimonadales | NA | NA |
| ASV1125 | 0.741 | 0.0001 | *** | | Cyanobacteria | Sericytochromatia | NA | NA | NA |
| ASV1403 | 0.740 | 0.0001 | *** | | Proteobacteria | Gammaproteobacteria | Betaproteobacteriales | Burkholderiaceae | NA |
| ASV537 | 0.736 | 0.0001 | *** |  | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | NA |
| ASV1387 | 0.732 | 0.0001 | *** |  | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Taibaiella sp. |
| ASV857 | 0.722 | 0.0001 | *** |  | Proteobacteria | Deltaproteobacteria | Bdellovibrionales | Bacteriovoracaceae | Chitinophaga sp. |
| ASV345 | 0.687 | 0.0006 | *** |  | Proteobacteria | Alphaproteobacteria | Rhizobiales | Rhizobiaceae | Peredibacter sp. |
| ASV1802 | 0.680 | 0.0016 | ** |  | Proteobacteria | Deltaproteobacteria | Bdellovibrionales | Bdellovibrionaceae | Rhizobium sp. |
| ASV70 | 0.664 | 0.0002 | *** |  | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Niastella sp. |
| ASV552 | 0.663 | 0.0002 | *** | | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | NA |
| TifTuf | |||||||||
| ASV1059 | 0.557 | 0.0051 | ** | | NA | NA | NA | NA | NA |
| ASV520 | 0.494 | 0.0047 | ** | | NA | NA | NA | NA | NA |
| ASV4025 | 0.490 | 0.0040 | * | | Dependentiae | Babeliae | Babeliales | Vemiphilaceae | NA |
| ASV796 | 0.475 | 0.0391 | * | | Bacteroidetes | Bacteroidia | Chitinophagales | Chitinophagaceae | Dinghuibacter sp. |
| ASV3054 | 0.454 | 0.0377 | * | Patescibacteria | Gracilibacteria | NA | NA | NA | |
| ASV2390 | 0.450 | 0.0362 | * | | Proteobacteria | Alphaproteobacteria | NA | NA | NA |
| ASV4 | 0.445 | 0.0091 | ** | NA | NA | NA | NA | NA | |
| ASV1605 | 0.428 | 0.0305 | * | | Patescibacteria | Gracilibacteria | Candidatus Perengrinibacteria | NA | NA |
| ASV174 | 0.426 | 0.0409 | * |  | NA | NA | NA | NA | NA |
| ASV2537 | 0.419 | 0.0447 | * | NA | NA | NA | NA | NA | |
| Leaves Roots Soil Latitude 36 TifTuf | |||||||||
| Fungi | % nt Identity | Accession | ASV | % Match | Latitude 36 | TifTuf |
|---|---|---|---|---|---|---|
| Fusarium concolor | 99.91% | GQ505763.1 | ASV52 | 100% | <0.1% | 1.10 ± 1.09% |
| Fusarium fujikuroi | 99.19% | CP023090.1 | ASV1344 | 1 nt mismatch | <0.1% | < 0.1% |
| Fusarium proliferatum | 99.10% | LS422791.1 | ASV1344 | 100% | <0.1% | < 0.1% |
| Cochliobolus lunatus | 99.04% | GQ221854.1 | ASV256 | 100% | <0.1% | < 0.1% |
| Aspergillus niger | 99.34% | KJ365316.1 | - | - | - | - |
| Nigrospora sphaerica | 98.36% | KT462720.1 | ASV1535 | 100% | <0.1% | <0.1% |
| uncultured Pelosporales | 96.43% | HG995490.1 | ASV410 | 100% | <0.1% | <0.1% |
| unknown Pleosporales sp. | 99.32% | JQ388930.1 | ASV92 | 100% | <0.1% | 0.25 ± 0.35% |
| uncultured Pleosporales | 97.27% | HG996374.1 | ASV36 | 100% | 0.65 ± 1.73% | 0.41 ± 0.76% |
| Bacteria | ||||||
| Bacillus velezensis | 98.72% | CP041361.1 | ASV2563 | 100% | <0.1% | <0.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, C.J.; Valiente, J.; Schiavon, M.; Dhillon, B.; Crow, W.T.; Stingl, U. Bermudagrass Cultivars with Different Tolerance to Nematode Damage Are Characterized by Distinct Fungal but Similar Bacterial and Archaeal Microbiomes. Microorganisms 2022, 10, 457. https://doi.org/10.3390/microorganisms10020457
Choi CJ, Valiente J, Schiavon M, Dhillon B, Crow WT, Stingl U. Bermudagrass Cultivars with Different Tolerance to Nematode Damage Are Characterized by Distinct Fungal but Similar Bacterial and Archaeal Microbiomes. Microorganisms. 2022; 10(2):457. https://doi.org/10.3390/microorganisms10020457
Chicago/Turabian StyleChoi, Chang Jae, Jacqueline Valiente, Marco Schiavon, Braham Dhillon, William T. Crow, and Ulrich Stingl. 2022. "Bermudagrass Cultivars with Different Tolerance to Nematode Damage Are Characterized by Distinct Fungal but Similar Bacterial and Archaeal Microbiomes" Microorganisms 10, no. 2: 457. https://doi.org/10.3390/microorganisms10020457