3.1. A General Survey of the Phylogeny of Alphaproteobacteria
Alphaproteobacteria constitute a large class of phenotypically diverse prokaryotes that has been divided in eight major orders and many families [
2,
5], predominantly with the conventional analysis of their 16S rRNA and phenotypic properties [
1,
2,
9,
13]. To evaluate and graphically render the phylogeny of these taxonomic divisions we have routinely used NuoL, the largest membrane subunit of complex I [
22,
25], as a single, informative protein marker, for the reasons presented in Materials and Methods.
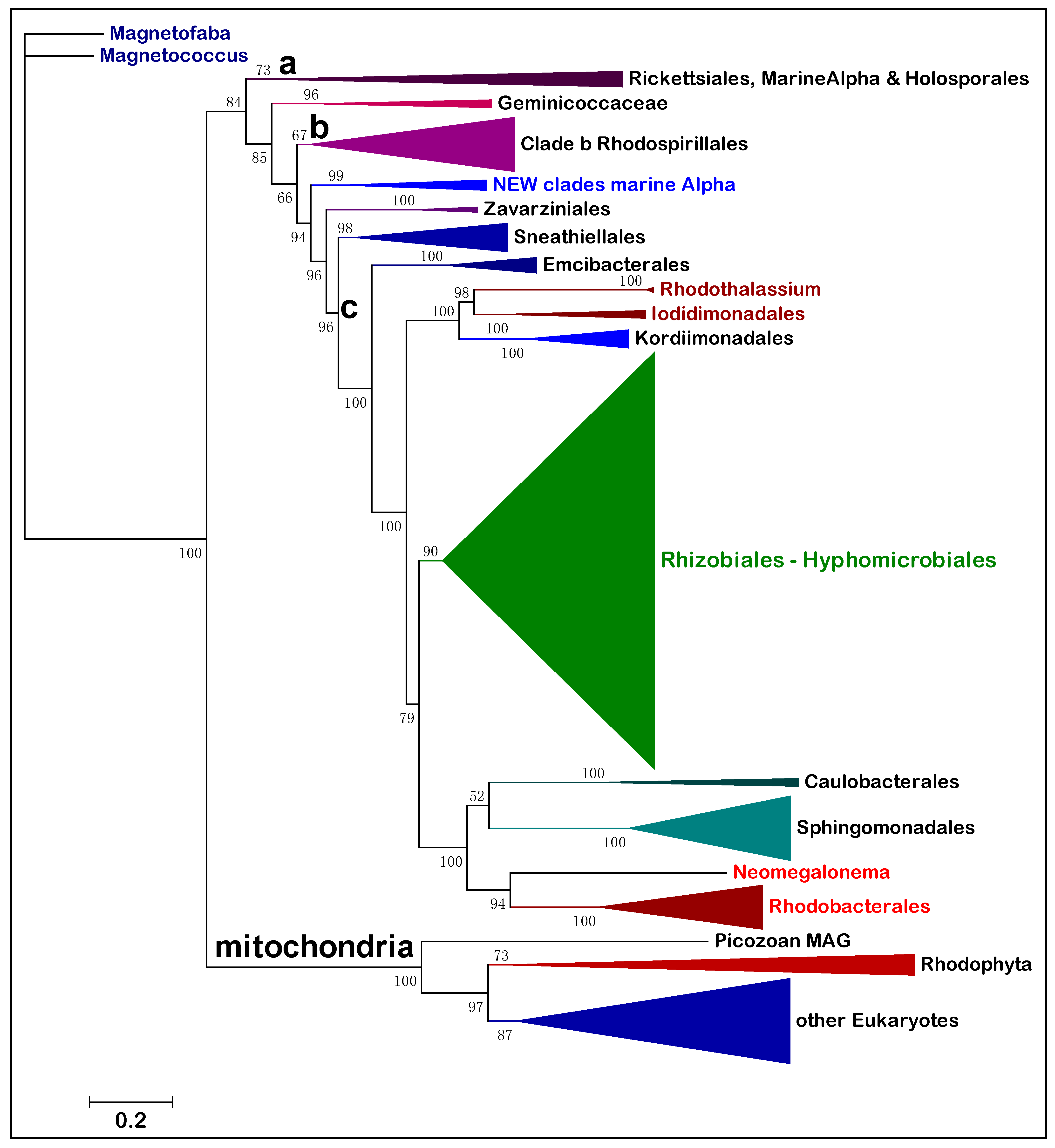
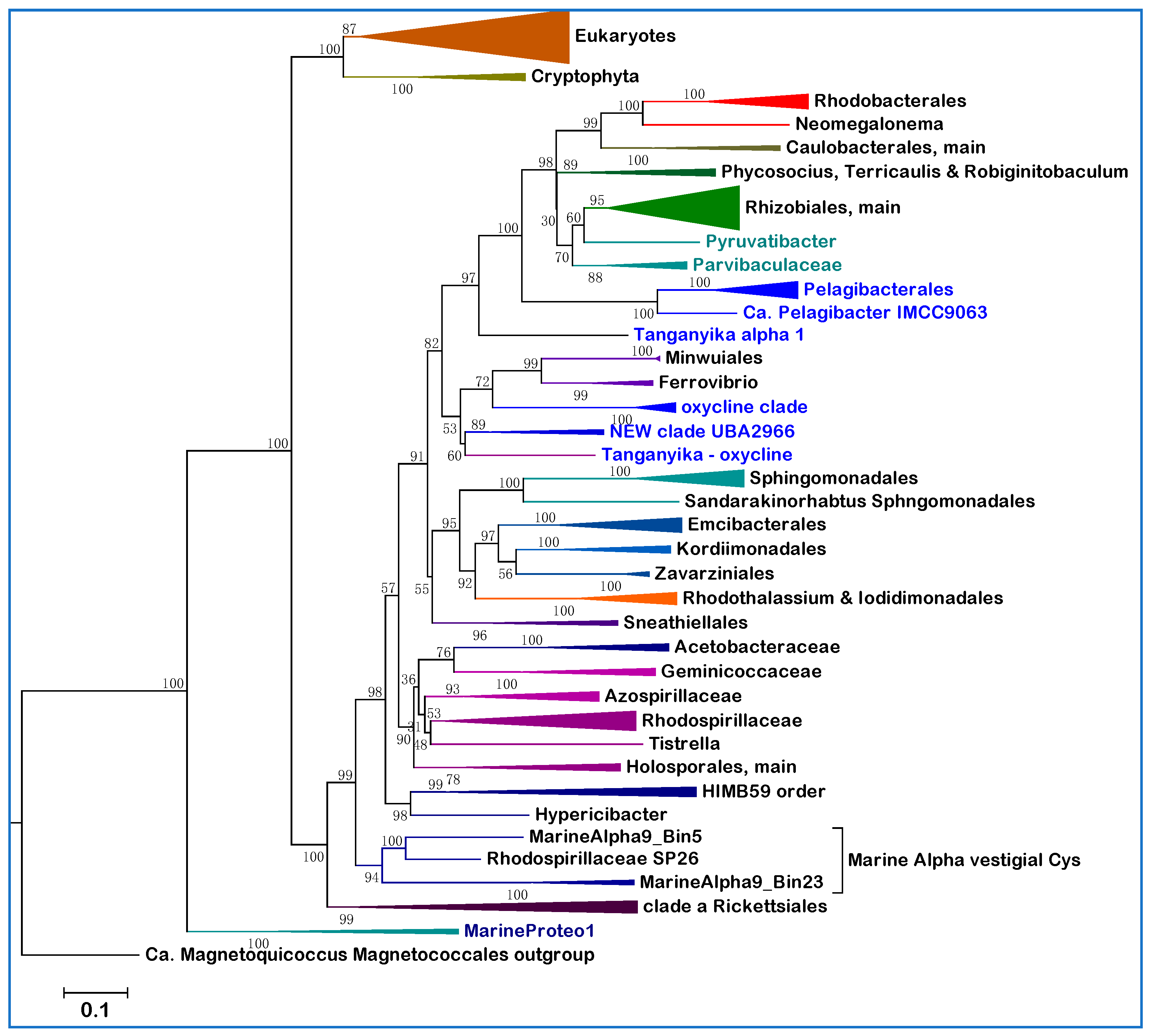
Figure 1 shows a representative ML tree obtained with a very broad set of NuoL proteins from Alphaproteobacteria and Eukaryotes. With few exceptions, notably Pelagibacterales as in previous studies [
13], these proteins represent all major orders and families in which Alphaproteobacteria are currently classified, thereby providing a graphical view of the relative abundance and phylogenetic diversity of taxa currently affiliated with the class. The tree is rooted on the distant NuoL homologs of Magnetococcales, generally considered the basal lineage of Alphaproteobacteria [
2,
4,
5,
13], and shows all the other bacterial lineages in a single clade that is sister to that of the mitochondrial ND5 homologs.
The tree in
Figure 1 shows an ‘Alphaproteobacteria-sister’ to mitochondria topology that is equivalent to that initially reported using 24 concatenated proteins, including NuoL and many marine MAGs [
11]. The same clustering has been recently reproduced with different taxonomic samplings of either Alphaproteobacteria or mitochondria [
26,
33]. However, the most common topology of phylogenetic trees combining Alphaproteobacterial and mitochondrial proteins shows the mitochondrial clade nested within the early branching part of the trees, often in a sister position to the Rickettsiales order [
2,
4,
11,
26]. Leaving aside the interpretation of these different topologies—see [
11,
24,
26] for a detailed discussion of the topic—the tree in
Figure 1 shows a fundamental feature relevant to the overall phylogeny of Alphaproteobacteria that has been scarcely noted before. The branching order of the various lineages follows a sequence of three major clades, which are labeled
a,
b and
c here. Clade
a includes the order of Rickettsiales and appears to be closely related to MAGs that have similarly low G+C content in their genomes, such as those affiliated to the TMED109 order of GTDB taxonomy [
11,
14]. Clade
b includes most Rhodospirillales and is generally separated from clade
a by the deep branching family [
13] or order [
12] of Geminicoccaceae/ales (
Table 1). Then, a series of minor orders of predominantly marine taxa, such as Sneathiellales, separates clade
b from clade
c, which includes all major lineages of Alphaproteobacteria, from Rhizobiales to Rhodobacterales (
Figure 1). An equivalent three-clades sequence can be discerned in the branching order of phylogenetic trees reported earlier, for example in studies on Alphaproteobacteria only [
3,
5,
13], as well as in studies focusing on the Alphaproteobacterial ancestry of mitochondria [
11,
26,
33]. Indeed, clade
c fundamentally corresponds to the ‘Core alphaproteobacteria’ group reported by Martijn et al., 2018 [
11].
The branching order of clade
b and
c differs from that of
Figure 1 in some recent studies, in which these major branches appear as sister clades [
5,
11,
26,
33]. The probable reason for this difference is the limited taxonomic sampling of Alphaproteobacteria in those studies, since phylogenetic trees reconstructed with much larger samplings [
13,
14] do show clade
b branching before clade
c as in
Figure 1. ML trees reconstructed with other single marker proteins, such as COX3 and NuoD, show a similar branching pattern to that in
Figure 1 too (see later
Section 3.5). Moreover, the lineages intermixed between clade
b and
c (
Figure 1) form a constant feature in phylogenetically broad trees including all such lineages [
13,
14]. Only Sneathiellales have been regularly included in other studies [
5,
11,
26], while single representatives of Kordiimonadales and Iodidimonadales have been shown to cluster with Sphingomonadales in clade
c [
11]. This clustering most likely derives from limited taxonomic samplings of the lineages forming the clade, a common distortion in phylogenic trees [
10,
13,
20]. Here we show that Sneathiellales, Kordiimonadales and other lineages at the base of clade
c present metabolic features that are essentially intermediate between those of representatives of clade
b and clade
c, sustaining the concept that Alphaproteobacteria have progressively evolved from anaerobic or facultatively anaerobic ancestral taxa of Rhodospirillales [
4,
10] to strictly aerobic taxa of marine Caulobacterales and Rhodobacterales [
4,
13,
21]. The deep divide between clade
b and
c in phylogenetic trees, therefore, may derive from a major evolutionary event related to the consolidation of permanent levels of oxygen in primordial earth [
21,
23], which enabled novel aerobic traits to arise in bacteria [
4,
21]. Traits, such as methanotrophy and aerobic nitrification, arose first in ancestors of extant Delta- and Gamma-proteobacteria and subsequently permeated the genome of Alphaproteobacteria by waves of Lateral Gene Transfer (LGT), presumably after some Rhodospirillales had already diverged [
27]. We introduce here the SERIK group to further corroborate this hypothetical evolutionary scenario, which derives from the phylogeny of Alphaproteobacteria (
Figure 1) combined with other considerations [
4,
10,
21,
27].
3.2. The SERIK Group and New Marine Clades of Alphaproteobacteria
SERIK is an acronym derived from the initial letters of Sneathiellales, Emcibacterales, Rhodothalassiales, Iodidimonadales and Kordiimonadales, following their branching sequence in descending phylogenetic trees, such as that presented in
Figure 1. These orders include almost exclusively marine taxa that have been introduced among Alphaproteobacteria in the last decade or so [
34,
35,
36,
37]. They have been validated as separate orders, clustering together in a single branch often labeled EKRS (i.e., in alphabetical order), by the recent re-classification of Alphaproteobacteria [
13], which has been adopted almost entirely in the current NCBI taxonomy system (
Table 1). However, only Sneathiellales is considered a separate order in the GTDB system, in agreement with [
5], classifying Emcibacterales, Rhodothalassiales, Iodidimonadales and Kordiimonadales in three families of the large order of Sphingomonadales (
Table 1). One of such families, the Rhodothalassiaceae, has been introduced earlier [
35], but is considered part of the separate order of Rhodothalassiales in other classifications [
13]. In GTDB taxonomy, the family Rhodothalassiaceae also includes the order Iodidimonadales (
https://gtdb.ecogenomic.org/searches?s=gt&q=f__Rhodothalassiaceae—accessed on 11 January 2021). Alphaproteobacterium Q-1, which is a consolidated member of the Iodidimonadales [
34,
36], is photosynthetic as
Rhodothalassium [
35]. However, the photosynthesis trait is absent in all other taxa of the SERIK group; therefore, it likely derives from sporadic LGT of the genetic island carrying all the genes required for photosynthesis, which is frequently exchanged in marine Rhodobacterales [
8,
9].
The different classification of SERIK lineages in NCBI and GTDB taxonomies (
Table 1) prompted a detailed phylogenomic analysis of uncultured bacteria that could be associated with SERIK taxa. The search focused on MAGs derived from recent metagenomes of marine environments (
Table 2) and was integrated with the phylogenetic analysis of major subdivisions of GTDB taxonomy that cluster together with the Sneathiellales in ML trees [
14]. In such trees, the branch containing the Sneathiellales includes the clade of Minwuiales, another minor marine order reported to be distinct from other bacteria [
37], plus two subdivisions of marine MAGs considered as separate orders (
Table 1): UBA2966, with the representative Rhodospirillaceae ARS1032, and GCA_002731375, named after the genome assembly code of Rhodospirillaceae NP113. Both these MAGs were found by the Tara Oceans Consortium [
38] and are represented in the MarineOcean DNA survey [
15], as well as in the metagenome of the Saanich Inlet [
18], a seasonally anoxic fjord that has been studied as a model for marine oxygen minimum zones (OMZ) [
17] (
Table 2).
Although representatives of these orders are not present in the metagenomes of many freshwater environments [
39], related taxa have been found in the anoxic depths of Lake Tanganyika, a unique stratified environment of deep freshwater [
40] (
Figure 2a). The NuoL tree of
Figure 1 includes a representative of Lake Tanganyika metagenome and three MAGs classified in the UBA2966 order, which form a distinct branch lying between clade
b and the SERIK group in NuoL trees (
Figure 2a). Seven MAGs from Lake Tanganyika have been classified as Sneathiellales using GTDB [
40], but Sneathiellales are marine-specific bacteria [
13,
41]. Considering their phylogenetic position and other features described below, such taxa of Lake Tanganyika are proposed to be part of novel clades of Alphaproteobacteria that may be characteristic of aquatic environments with steep oxygen gradients; they are cumulatively called new clades here. Alphaproteobacteria MarineAlpha1_Bin1 appears to constitute a precedent for such new clades, since it was reported to lie basal to the equivalent of clade
c in several phylogenetic trees [
11,
42] and its NuoL protein clusters with the oxycline clade overlapping o_GCA_002731375 (
Figure 2a). The genome of MarineAlpha1_Bin1 is of very low quality and has only a fraction of the set of ‘alphamitoCOGs’ used by Ettema and coworkers to construct phylogenetic trees [
11,
42]. Yet, such trees invariably positioned this MAG away from other marine MAGs, except for MarineAlpha2_Bin1, which belongs to another GTDB order, o_GCA-2701885, annotated in
Table 2. Bioenergetic proteins of this order do not segregate with the same oxycline clade as those of MarineAlpha1_Bin, but close to members of the UBA2966 clade and one MAG from Lake Tanganyika (
Supplementary Figure S2).
3.3. The New Marine Clades Appear to Cluster with Sneathiellales but Have Different Functional Traits
Given their original classification, the Alphaproteobacterial taxa that form part of the new clades introduced here could belong to either the Rhodospirillales ‘super-order’ or the Sneathiellales order. To discriminate between these possibilities, we produced alignments of single marker proteins from representatives of most subdivisions of Rhodospirillales in GTDB taxonomy (
Table 2) combined with those of selected taxa of the new clades and of Sneathiellales [
14].
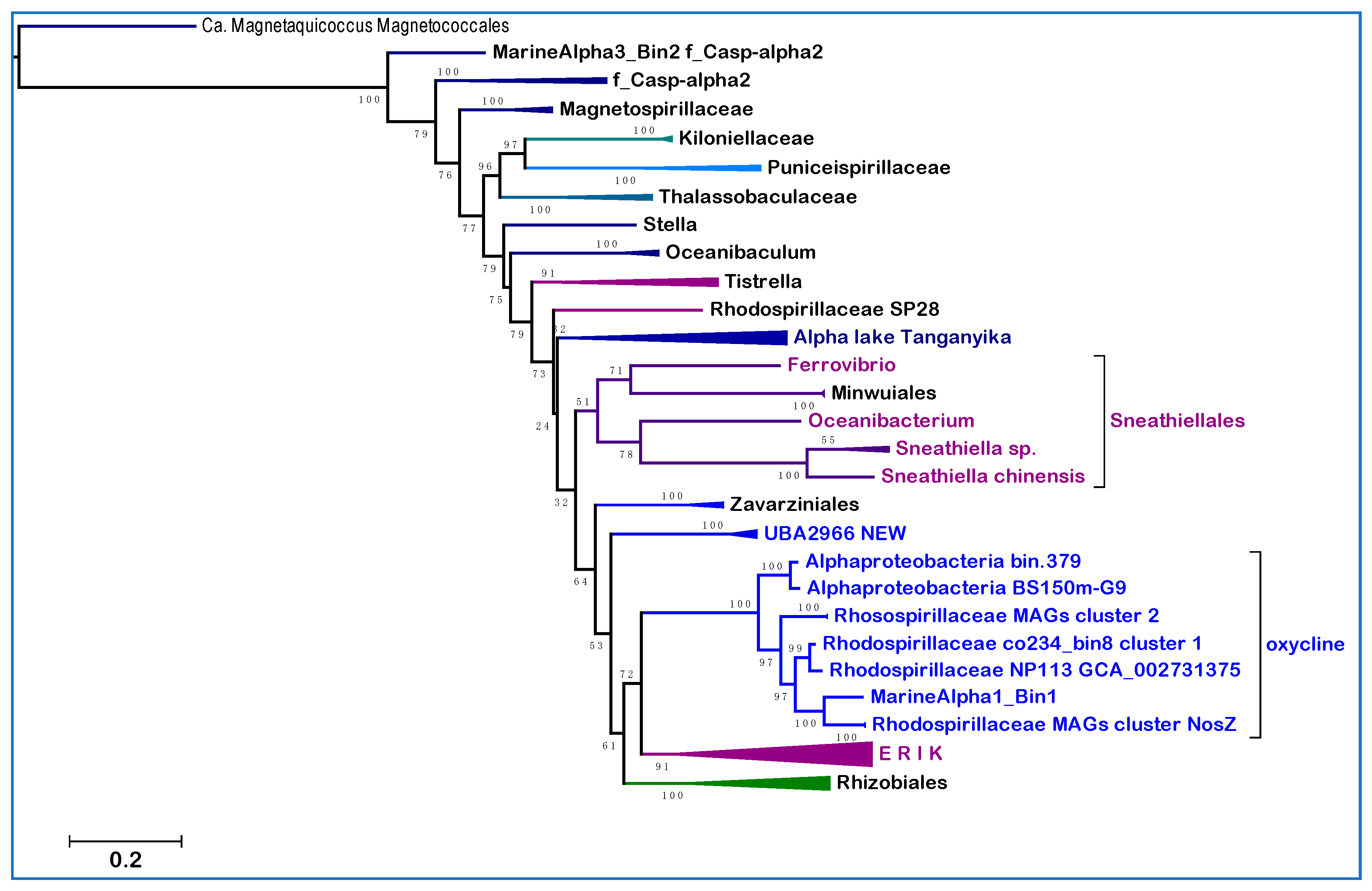
Figure 2a shows a typical tree obtained with the NuoL protein indicating that MAGs of the new clades cluster with representatives of either the UBA2966 or the GCA_002731375 order of GTDB taxonomy, away from taxa classified in NCBI taxonomy but within the large clade that includes all cultivated Sneathiellales, such as
Oceanibacterium and Minwuiales (
Figure 2a). Proteins of Lake Tanganyika taxa classified as Sneathiellales [
40] form the earliest branches of the clade, which lies in a sister position to the clade, including most proteins from Rhodospirillales (
Figure 2a). Although the bootstrap support for the separation of these large clades is not very high, the tree topology in
Figure 2a has been reproduced in multiple trees with different models and taxonomic samplings. Since the statistical weight of consistent bifurcating topologies obtained in separate trees is much stronger than branch bootstrap support [
20], the separation of Sneathiellales from Rhodospirillales likely reflects a fundamental split in the phylogeny of Alphaproteobacteria, as indicated by much broader trees (see
Figure 1 and previous discussion at the end of
Section 3.1). Given the clustering of the members of the new clades with Sneathiellales (
Figure 2a), it appears that such clades may represent families or sub-orders related to the Sneathiellales order. Consequently, their original denomination of ‘Rhodospirillaceae bacterium’ [
18] should be rectified.
Nevertheless, subsequent phylogenetic (
Figure 3) and functional analysis (
Figure 2b) suggest that the new clades should be considered taxonomically separate from Sneathiellales. The results of our detailed analysis of functional traits linked to aerobic metabolism, summarized in
Figure 2b, strongly sustains this possibility. The first piece of evidence regards Q and the reoxidation of its reduced form, ubiquinol, catalyzed by ubiquinol-cytochrome
c reductase (complex III), which is present in all the taxa in
Figure 2b and the great majority of their relatives. Some Sneathiellales can additionally oxidize ubiquinol using low concentrations of oxygen because they possess one or more cytochrome
bd ubiquinol oxidases of the CIO type [
21,
27]—top left of
Figure 2b. However, members of the new clades and associated MAGs of Lake Tanganyika do not have such alternative oxidases to discharge reduced Q; yet they have the capacity of synthesizing Q also under anoxic conditions, since their genome often contains the
ubiTUV cluster that is required for this anaerobic biosynthesis [
29] (
Figure 2b). Genes for ubiTUV proteins are present in Magnetococcales and in a few Rhodospirillales [
29], but otherwise are rare in marine Alphaproteobacteria, as verified by detailed genomic analysis. Therefore, their presence in members of the new clades (
Figure 2b) indicates an uncommon adaptive trait to anoxic conditions and implies a redox overload on
c-type cytochromes by ubiquinol-cytochrome
c reductase. The second piece of evidence regards the discharge of this redox overload, which is carried out by an uncommon variety of terminal oxidases of the HCO superfamily (
Figure 2b, cf. [
21]).
All families of HCO are present in Alphaproteobacteria, but infrequently together in the same genome [
21,
27]. Soil Rhizobiales of the
Bradyrhizobium genus are an exception, since they have genes for members of HCO family A, B and C in their genome, often with different subtypes of A family COX, such as
ccoNOPQ [
43], corresponding to subtype a-III [
21,
27]. Although hard evidence is lacking, subtype a-III is likely to operate at lower oxygen concentrations than the main subtype b COX [
21,
27,
43]. Among marine Alphaproteobacteria, COX operon subtype a-III is present in clades of Pelagibacterales living near or within OMZ [
44] and a few Rhodospirillales, for example
Inquilinus [
21]. Remarkably, they are concentrated in the MAGs of the new clades, together with C family oxidases and also a specific subtype of B family oxidases, previously labeled ba3-a1 [
4,
21] (
Figure 2b). Hence, the genomes of the new clades of Alphaproteobacteria introduced here have an exceptional combination of three families of HCO terminal oxidases, which clearly distinguishes them from Sneathiellales and other SERIK members.
Before describing further phylogenetic analysis, it is worth discussing the N cycle [
30], another functional trait that is characteristic of the new clades (
Figure 2b). Recently, a novel element of this cycle has been identified in the HCO subtype ba3-a1 [
21], which has been found to function as a nitric oxide reductase using reduced cytochrome
c as electron donor [
45], similarly to the classical nitric oxide reductase of the N cycle, NOR [
17,
30,
46,
47]. The ba3-a1 with NOR-like activity is widespread among members of both the oxycline and NEW—UBA2966 clades (
Figure 2b), while only
Minwuia has classical NOR among the taxa shown in
Figure 2b. Several taxa have other enzymes of the N cycle, including Nitrous Oxide (N
2O) Reductase, usually labeled NosZ after the
nosZ gene of its catalytic protein [
30] (
Figure 2b). However, only one representative of each clade is potentially capable of undertaking complete aerobic denitrification, similarly to
Minwuia: Alphaproteobacteria BS150m-G9 of the oxycline clade, as well as Alphaproteobacteria SIO54-bin61 of NEW—UBA2966. These two MAGs have the cd1 type of nitrite reductase as their relatives and would use ba3-a1 as a NOR to provide the substrate for their NosZ, which clusters in a different clade than that of
Minwuia in phylogenetic trees (
Supplementary Figure S3). However, the majority of taxa forming the new clades introduced here do not have a functional NosZ and therefore would contribute to N
2O accumulation in their marine environment [
17,
46,
48].
Figure 2.
Phylogeny and functional profile of new clades of Alphaproteobacteria. (
a) The phylogenetic ML tree was reconstructed with the LG model using NuoL proteins from Rhodospirillales and Sneathiellales taxa labeled according to GTDB taxonomy and the clades introduced here (
Table 1; * Magnetovibrionaceae include also f_Casp-alpha; the bracket embraces all taxa of Sneathiellales); (
b) metabolic traits and genomic properties of Sneathiellales and representatives of the clades introduced here. ISP stands for the Iron Sulfur Protein subunit of ubiquinol-cytochrome
c reductase; the orange boxes indicate the presence of the insert typical of Rhodospirillales that separates the conserved stretches with the Fe-S ligands [
27]. SCO (Synthesis of Cytochrome C Oxidase) is a COX accessory protein involved in copper insertion which is often associated with B family HCO [
4]. Terminal oxidases include CIO (Cyanide Insensitive Oxidase) of the cytochrome
bd ubiquinol oxidases [
27], which is present only in some Sneathiellales, and one or two HCO A family subtypes, aa3 b and a-III [
4,
21]. Cbb3 identifies the complete operon of C family HCO [
21]. *Subtype ba3-a1 of B family HCO [
4] was suspected to be involved in nitrogen metabolism (see [
21] and references therein) and has been recently reported to exhibit NO reductase activity, hence re-named eNOR [
45]. It is thus part of the N cycle, which is usually incomplete in marine biomes because of the limited distribution of classical NOR (NO reductases) and NosZ (N
2O reductases) [
17,
30]. Clade I, TAT-dependent nitrous oxide reductase [
17], is annotated as NosZ, while only
Minwuia has a classical cytochrome
c-reducing NOR completing aerobic denitrification [
30]. See
Supplementary Figure S3 for the taxonomic distribution of NosZ. Nitrite (NO
2) reductases are defined according to their structure (cd1) or gene (NirK for the Cu-dependent and NirBD for the NAD(P)H-dependent) [
30]. Except one
Sneathiella, which has transmembrane Nar, all taxa shown have the NapA type of periplasmic nitrate (NO
3) reductase [
30]. The boxes are colored with increasing intensity when more than one gene/operon is present in the genome. The proteins for Q biosynthesis reported on the right are annotated according to their genes [
29]: t for sterol-carrier-like
ubiT, u and v for
ubiU and
ubiV that have a U32 peptidase fold; q9 for CoQ9, an accessory protein shared by Alphaproteobacteria and mitochondria [
4]. The last column on the right (%GC) lists the percentage of GC bases in the genomes of the taxa, a parameter strongly correlated with taxonomic relatedness in most Alphaproteobacteria [
13]. Here it is shown to vary considerably among Sneathiellales genera, while remaining close to the average value of 59% for the MAGs of the oxycline clade. Note that the classification of Lake Tanganyika MAGs is very tentative.
Figure 2.
Phylogeny and functional profile of new clades of Alphaproteobacteria. (
a) The phylogenetic ML tree was reconstructed with the LG model using NuoL proteins from Rhodospirillales and Sneathiellales taxa labeled according to GTDB taxonomy and the clades introduced here (
Table 1; * Magnetovibrionaceae include also f_Casp-alpha; the bracket embraces all taxa of Sneathiellales); (
b) metabolic traits and genomic properties of Sneathiellales and representatives of the clades introduced here. ISP stands for the Iron Sulfur Protein subunit of ubiquinol-cytochrome
c reductase; the orange boxes indicate the presence of the insert typical of Rhodospirillales that separates the conserved stretches with the Fe-S ligands [
27]. SCO (Synthesis of Cytochrome C Oxidase) is a COX accessory protein involved in copper insertion which is often associated with B family HCO [
4]. Terminal oxidases include CIO (Cyanide Insensitive Oxidase) of the cytochrome
bd ubiquinol oxidases [
27], which is present only in some Sneathiellales, and one or two HCO A family subtypes, aa3 b and a-III [
4,
21]. Cbb3 identifies the complete operon of C family HCO [
21]. *Subtype ba3-a1 of B family HCO [
4] was suspected to be involved in nitrogen metabolism (see [
21] and references therein) and has been recently reported to exhibit NO reductase activity, hence re-named eNOR [
45]. It is thus part of the N cycle, which is usually incomplete in marine biomes because of the limited distribution of classical NOR (NO reductases) and NosZ (N
2O reductases) [
17,
30]. Clade I, TAT-dependent nitrous oxide reductase [
17], is annotated as NosZ, while only
Minwuia has a classical cytochrome
c-reducing NOR completing aerobic denitrification [
30]. See
Supplementary Figure S3 for the taxonomic distribution of NosZ. Nitrite (NO
2) reductases are defined according to their structure (cd1) or gene (NirK for the Cu-dependent and NirBD for the NAD(P)H-dependent) [
30]. Except one
Sneathiella, which has transmembrane Nar, all taxa shown have the NapA type of periplasmic nitrate (NO
3) reductase [
30]. The boxes are colored with increasing intensity when more than one gene/operon is present in the genome. The proteins for Q biosynthesis reported on the right are annotated according to their genes [
29]: t for sterol-carrier-like
ubiT, u and v for
ubiU and
ubiV that have a U32 peptidase fold; q9 for CoQ9, an accessory protein shared by Alphaproteobacteria and mitochondria [
4]. The last column on the right (%GC) lists the percentage of GC bases in the genomes of the taxa, a parameter strongly correlated with taxonomic relatedness in most Alphaproteobacteria [
13]. Here it is shown to vary considerably among Sneathiellales genera, while remaining close to the average value of 59% for the MAGs of the oxycline clade. Note that the classification of Lake Tanganyika MAGs is very tentative.
![Microorganisms 10 00455 g002]()
3.4. Functional and Phylogenetic Diversity of the Oxycline Clade
The analysis of the traits presented in
Figure 2b suggests that two of the clades of marine Alphaproteobacteria introduced here, namely the NEW—UBA2966 and the ‘oxycline clade’ (
Table 2), clearly differ from members of the Sneathiellales in the following functional characters: (1), the absence of ubiquinol oxidases; (2), the capacity of Q biosynthesis under anoxic conditions; (3), the rare combination of HCO terminal oxidases; (4), the presence of ba3-a1 oxidase that may function as NOR; and (5), the abundance of enzymes for aerobic denitrification. These functional diversities suggest a strong divergence from Sneathiellales, despite the clustering observed in trees with limited taxonomic sampling (
Figure 2a). We then focused our analysis on the ‘oxycline clade’ because the distribution of its distinctive functional traits is more compact than in the NEW—UBA2966 clade, some taxa of which do not have such traits (
Figure 2b). Moreover, proteins from taxa of Lake Tanganyika often intermix with those of NEW—UBA2966; therefore, this clade may have a higher taxonomic rank than the ‘oxycline clade’. We tentatively consider the latter clade as a family level taxonomic division encompassing the previously ill-defined GCA_002731375 order [
14] (see
Supplementary Table S1 for further details).
We used CTP synthase as a single protein marker to expand the phylogenetic analysis of the oxycline clade. This large soluble enzyme is part of the 120 protein markers used for GTDB taxonomy [
12] and reacts with nucleotides as the majority of the same proteins. In the absence of sequence data for the 16S RNA in MAGs [
17,
18,
19,
38], CTP synthase can provide a valuable marker for taxonomic classification of Alphaproteobacteria, having features complementary with those of NuoL [
10].
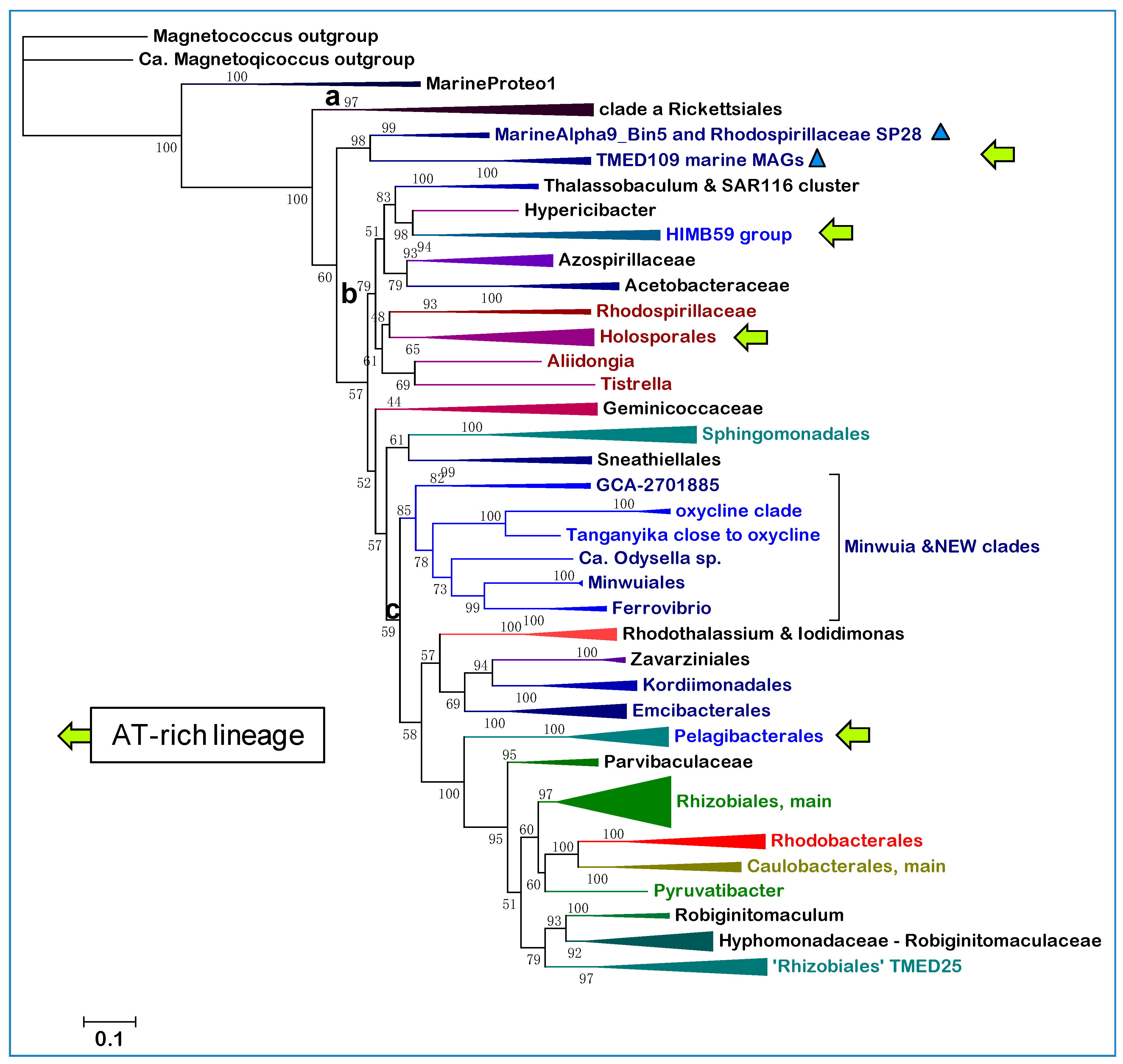
Figure 3 shows a representative ML tree of this protein from Alphaproteobacterial taxa spanning deep branching Rhodospirillales to early branching Rhizobiales, therefore encompassing the phylogenetic space of the new clades (
Figure 1). The oxycline clade is separate from the large clade of Sneathiellales, branching after the clade of Zavarziniales—a lineage originally associated with Acetobacteraceae, but later found to form a separate family [
13] or order [
12] (
Table 1). The NEW—UBA2966 clade lies intermediate between the Zavarziniales and the oxycline clade and together with the latter forms a sister branch to that, including the ERIK lineages (
Figure 3). This observation has been confirmed in trees obtained with much wider taxonomic sampling.
Figure 3.
Phylogeny of the oxycline clade using the CTP synthase marker. The ML tree was reconstructed with the LG model using the sequences of CTP synthase [
10] from an assortment of Rhodospirillales and Sneathiellales taxa similar to that used for the tree in
Figure 2a, plus various representatives of the ERIK orders of the SERIK group and some early branching Rhizobiales, for example
Parvibaculum. The alignment included 60 sequences with 568 amino-acid sites. The labelling of the various taxa of the oxycline clade follows the nomenclature shown in
Supplementary Table S1.
Figure 3.
Phylogeny of the oxycline clade using the CTP synthase marker. The ML tree was reconstructed with the LG model using the sequences of CTP synthase [
10] from an assortment of Rhodospirillales and Sneathiellales taxa similar to that used for the tree in
Figure 2a, plus various representatives of the ERIK orders of the SERIK group and some early branching Rhizobiales, for example
Parvibaculum. The alignment included 60 sequences with 568 amino-acid sites. The labelling of the various taxa of the oxycline clade follows the nomenclature shown in
Supplementary Table S1.
The tree in
Figure 3 additionally shows a granular definition of subdivisions within the oxycline clade, which tentatively include three major species-level clusters as detailed in
Supplementary Table S1. Notably, the CTP synthase of MarineAlpha1_Bin1 associates with full support to the cluster defined as NosZ for the presence of this trait in its MAGs (
Supplementary Table S1). Hence, MarineAlpha1_Bin1 can be considered a
bona fide member of the oxycline clade. Intriguingly, all the CTP synthase sequences of the oxycline clade show an insert of 10–12 residues that appear to be unique among Alphaproteobacteria, thereby constituting a molecular signature useful for classification.
Next, we enlarged the taxonomic breadth of our analysis, encompassing all known lineages of Alphaproteobacteria using different protein markers. Although the branching sequence of the oxycline clade and associated new clades was essentially confirmed, we noted their tendency to segregate with Minwuiales and
Ferrovibrio, while the core group of Sneathiellales branched in apparent sister position to the order Sphingomonadales, as reported earlier [
5,
11,
26,
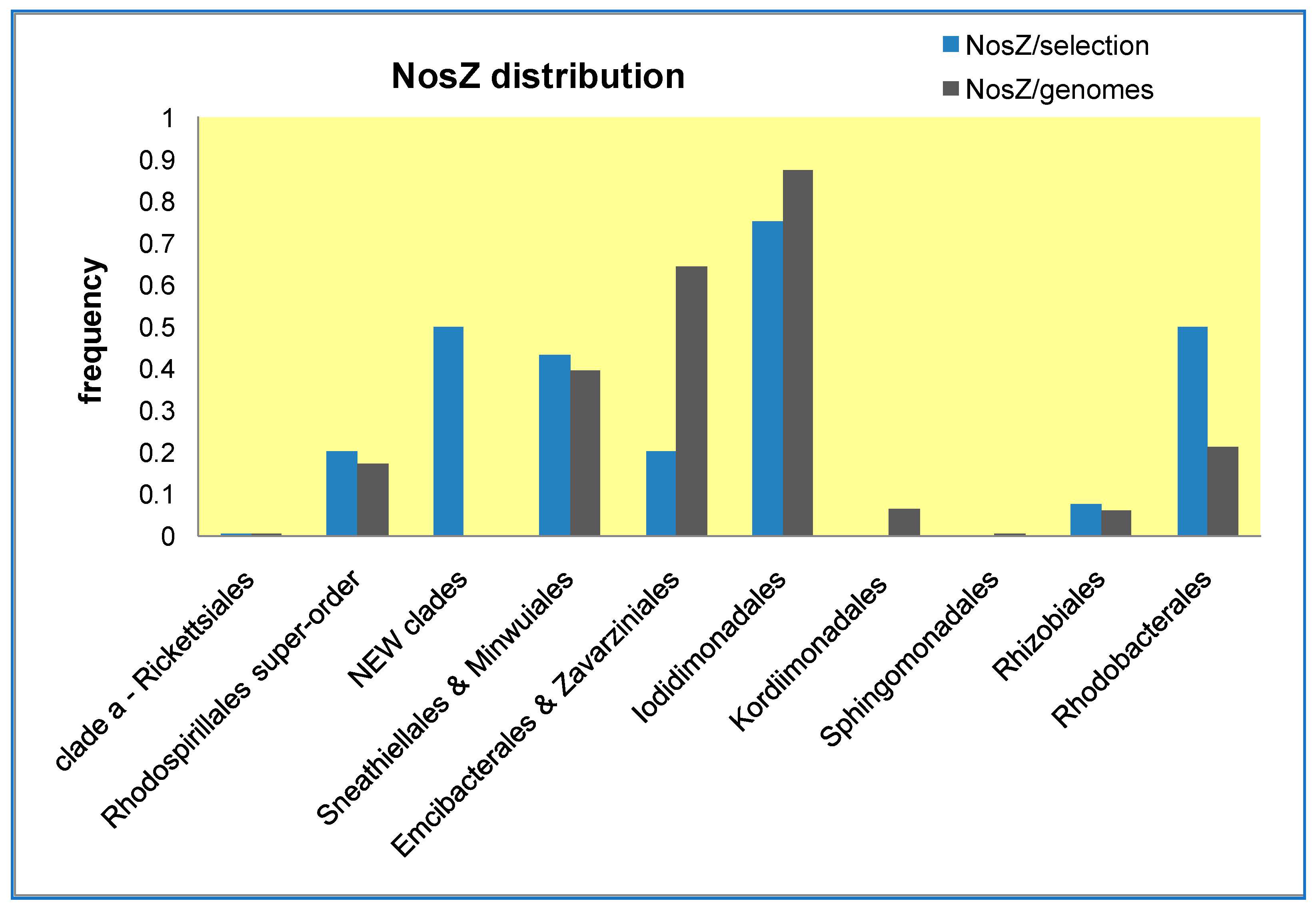
42]. To verify whether Sphingomonadales may be functionally related to Sneathiellales, we have analyzed the lineage-specific frequency of the gene encoding NosZ, the copper enzyme that completes the denitrification pathway [
30] (
Figure 4). Results obtained with two alternative approaches for evaluating the lineage-specific frequency of this gene show that Sphingomonadales are clearly distinct from Sneathiellales and other SERIK lineages, some of which, in particular Iodidimonadales, display the highest frequency (
Figure 4).
3.5. The Expanded Phylogeny of Alphaproteobacteria and Its Problems
As single phylogenetic markers, NuoL and CTP synthase suffer from compositional heterogeneity problems similar to those experienced when these proteins are concatenated with other markers in super-matrix alignments [
3,
5,
11,
12,
26]. It is generally considered that the major source of such problems is the strong bias towards GC-poor, and consequently AT-rich genomes in lineages with an endoparasitic lifestyle, such as Rickettsiales [
5,
26] and marine taxa with streamlined genomes: MarineAlpha9 [
11], HIMB59 [
3,
5,
11], and Pelagibacterales [
3,
11,
44]. The genome-derived compositional bias of coded proteins, combined with their high mutation rate [
5,
11,
26], produce phenomena of Long Branch Attraction (LBA) [
3,
4,
5] between AT-rich Alphaproteobacteria, and also between these bacteria and similarly AT-rich mitochondria [
3,
4,
5,
11,
24,
26,
33]. In the ML tree of
Figure 1, LBA has influenced the clustering of MarineAlpha9 and Holosporales within clade
a. Holosporales are either a separate order [
13,
14] or family [
5,
10] of obligate endocellular parasites that have been traditionally associated with Rickettsiales (see [
5] and References therein). However, ML trees reconstructed from NuoL alignments of smaller or different taxonomic samplings than that used in
Figure 1 do separate Holosporales from clade
a, clustering them within clade
b as it is now considered to be correct [
5,
26,
33]. Nevertheless, MarineAlpha9, HIMB59, and often Pelagibacterales, remain clustered with Rickettsiales in most NuoL trees, suggesting underlying LBA effects, most likely due to the bias towards hydrophobic amino acids in AT-rich genomes [
5,
26], which would inevitably affect a strongly hydrophobic membrane protein such as NuoL.
After analyzing various proteins shared by Alphaproteobacteria and mitochondria, we found that most LBA effects and compositional heterogeneity problems disappear in phylogenetic trees reconstructed from taxonomically broad alignments of the NuoD subunit of complex I (
Figure 5). Bacterial NuoD is a close homolog of Nad7 encoded by the mitochondrial DNA of Archaeplastida and other protists [
22,
31,
32], but is encoded in the nuclear DNA of other eukaryotes and labeled 49kDa subunit [
22,
25]. NuoD is a soluble protein and therefore is expected to be less affected by genome-induced compositional bias than transmembrane proteins such as NuoL. Moreover, it is very conserved, especially at the C-terminus where vestigial Cys ligands of the Ni-Fe cofactor that were present in its ancestor, the large catalytic subunit of hydrogenases, are sometimes retained [
22]. We found this to be the case in the NuoD sequences of MarineAlpha9 taxa and a few other marine Alphaproteobacteria that cluster with them (for example Rhodospirillaceae SP28,
Figure 5), consequently anchoring such taxa at the basis of Alphaproteobacterial phylogeny. Indeed, vestigial Cys ligands in NuoD were previously found only among deep branching bacteria [
22].
Detailed examination of the NuoD tree in
Figure 5 indicates that the proteins from AT-rich lineages segregate away from Rickettsiales, which remain the fundamental component of clade
a. Holosporales cluster in the middle of clade
b as in trees obtained after complex correction of concatenated alignments for compositional heterogeneity [
5]. HIMB59 and related taxa such as MarineAlpha5 cluster with another branch of clade
b, while Pelagibacterales cluster within clade
c, just before the separation of Rhizobiales and other major lineages of the class (
Figure 5). These branching positions are the same as those obtained after stationary-trimming of multiple protein alignments to sequentially remove site heterogeneity and reduce compositional bias [
11]. The branching order of Pelagibacterales is also equivalent to that reported in Bayesian or ML trees corrected with different procedures to limit LBA effects [
3,
26,
33]. Here, using a suitable single marker protein and wide taxonomic samplings, we have obtained phylogenetic trees that show the most appropriate segregation of different AT-rich genomes, as if we had applied sophisticated procedures to minimize LBA and compositional bias artefacts. Such procedures have been criticized because they inevitably reduce phylogenetic signal [
24], removing sites without considering their position in the structural architecture of the aligned proteins. Ultimately, this work shows that it is not necessary to apply sophisticated bioinformatic corrections for obtaining a comprehensive phylogenetic picture of all major lineages of Alphaproteobacteria. Therefore, that ‘the only way to counter artefacts [of this kind] is by removing compositionally biased sites, or taxa’ [
5] appears to be a bioinformatic myth. Conversely, the relatively short length and strong conservation of the NuoD protein (the alignment used for the tree in
Figure 5 has less than 500 amino acid positions) prevents discrete resolution of several branches of clade
c. These are undoubtedly better resolved in NuoL trees (
Figure 1), or with concatenated protein alignments (cf. [
11,
26]).
To reduce the problem of limited resolution in the branches of clade
c, the alignment used in
Figure 5 was extended to NuoD proteins of late branching lineages, such as Hyphomonadaceae and
Robiginitomaculum, which are now considered to be sisters rather than part of the related orders of Rhodobacterales and Caulobacterales [
13,
49]. The MAGs classified in the TMED25 division of Rhizobiales [
14], which are abundant in marine metagenomes (
Table 2 and [
15]), cluster together with Hyphomonadaceae and Robiginitomaculaceae (
Figure 5), as expected from the highly derived genomes of these taxa. On the other hand, Parvibaculaceae are shown to be the earliest branching family of Rhizobiales (
Figure 5), consistent with some studies [
6,
13], but apparently in contrast with another [
7].
3.6. Phylogenetic Trees of NuoD with Both Alphaproteobacteria and Mitochondria
Given the proven value of NuoD as a single phylogenetic marker for the phylogeny of Alphaproteobacteria (
Figure 5), it was natural to extend its alignment to homologous Nad7 proteins encoded by the mitochondrial DNA of protists [
22]. Representative ML trees, as that shown in
Figure 6, displayed the Alphaproteobacteria-sister topology to the mitochondrial clade as in
Figure 1 and previously reported trees [
11,
26,
33]. The tree in
Figure 6 is presented in ascending order for enabling its direct comparison with such published trees [
11,
26,
42]. It shows a decreased resolution of clade
c branches with respect to the tree obtained with Alphaproteobacterial proteins only (
Figure 5), also because of its smaller taxonomic sampling. Moreover, some LBA effects, presumably due to the inclusion of mitochondrial sequences, seem to have pulled the branch of the HIMB59 order in between clade
a and
b (
Figure 5 and
Figure 6). However, the separate tree position of major AT-rich lineages, such as Pelagibacterales, remains the same as in
Figure 5, without any clustering of Rickettsiales with mitochondria (
Figure 6). By contrast, clustering of mitochondria with Rickettsiales and Holosporales was regularly found in phylogenetic trees constructed with concatenated alignments of COX1-2-3 proteins, or single accessory proteins such as CtaG_Cox11 [
23]. Similar findings have been reported using concatenations of these and other bioenergetic proteins [
24].
Intriguingly, the addition of NuoD sequences from the early branching group of MarineProteo1 [
11] produces a new branch subtending the sister clades of mitochondria and all Alphaproteobacteria in NuoD trees (
Figure 6), as well as in NuoL trees. These results fundamentally agree with the increasing number of studies [
11,
26,
33,
42] supporting the hypothesis that the progenitors of mitochondria did not emerge from within the Alphaproteobacteria class, but from a related lineage of Proteobacteria [
11,
26]. The hypothesis derives from a strict interpretation of phylogenetic trees as that in
Figure 6 [
11,
26], assuming that such trees can capture the evolutionary information retained in the proteins of Alphaproteobacteria and mitochondria that are available today. However, this assumption is not guaranteed a priori, since the phylogenetic signal of an ancient transition, such as mitochondrial evolution, estimated to be around 1.8 billion years old [
21,
33], might have been lost almost entirely in contemporary proteins [
4,
20,
23,
26]. Combining together more and more proteins of different type and evolutionary history to reconstruct phylogenetic trees [
11,
26] does not necessarily enhance the residual phylogenetic signals that some of these proteins may have, producing instead additional sequence entropy that is difficult to disentangle from genuine molecular information of evolutionary value [
16,
20]. This can be obtained relatively easily by focusing on protein markers with a solid phylogenetic signal [
20,
50], such as NuoD (
Figure 5 and
Figure 6). Indeed, the classical single marker 16S RNA still produces valid phylogenetic information for the classification of Alphaproteobacteria, provided that exhaustive taxonomic samplings are used to reconstruct its trees [
13].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





