
Identification and Validation of Ikaros (IKZF1) as a Cancer Driver Gene for Marek’s Disease Virus-Induced Lymphomas

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Bird Experiments and Analyses
2.3. Tissue Collection and Further Processing
2.4. Bioinformatics
2.4.1. DNA Sequencing
2.4.2. Detection of Somatic Single-Nucleotide Variants (SNVs) and Small Insertions and Deletions (Indels)
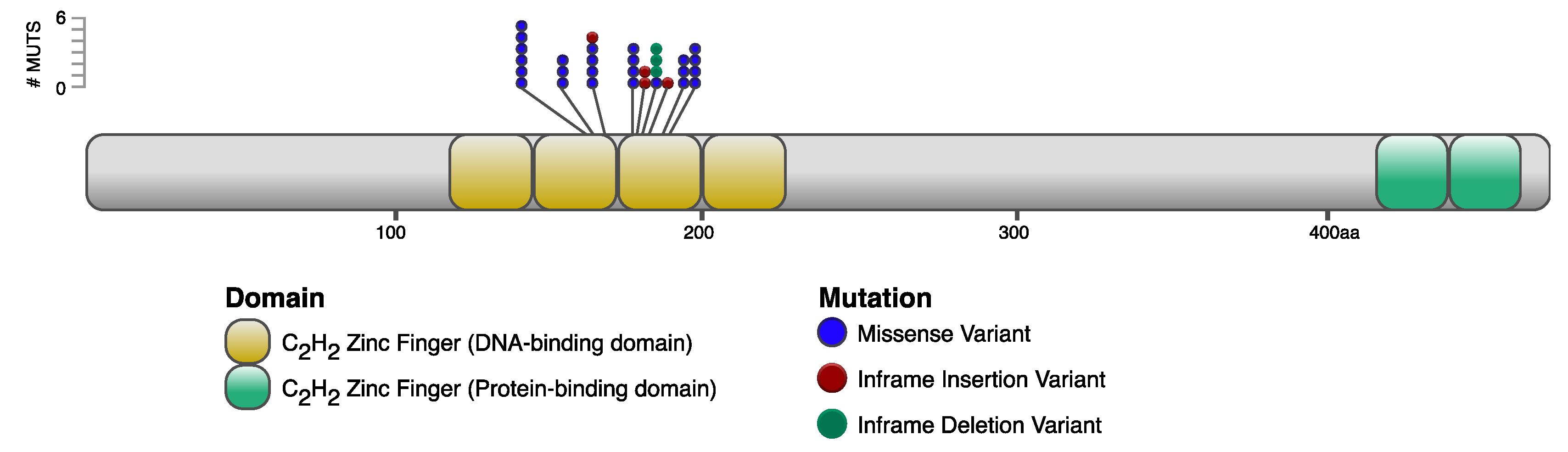
2.4.3. Annotation of IKZF1 Variants on Ikaros Protein Domains
3. Results
3.1. MD Tumors Contain Key Somatic Mutations in IKZF1
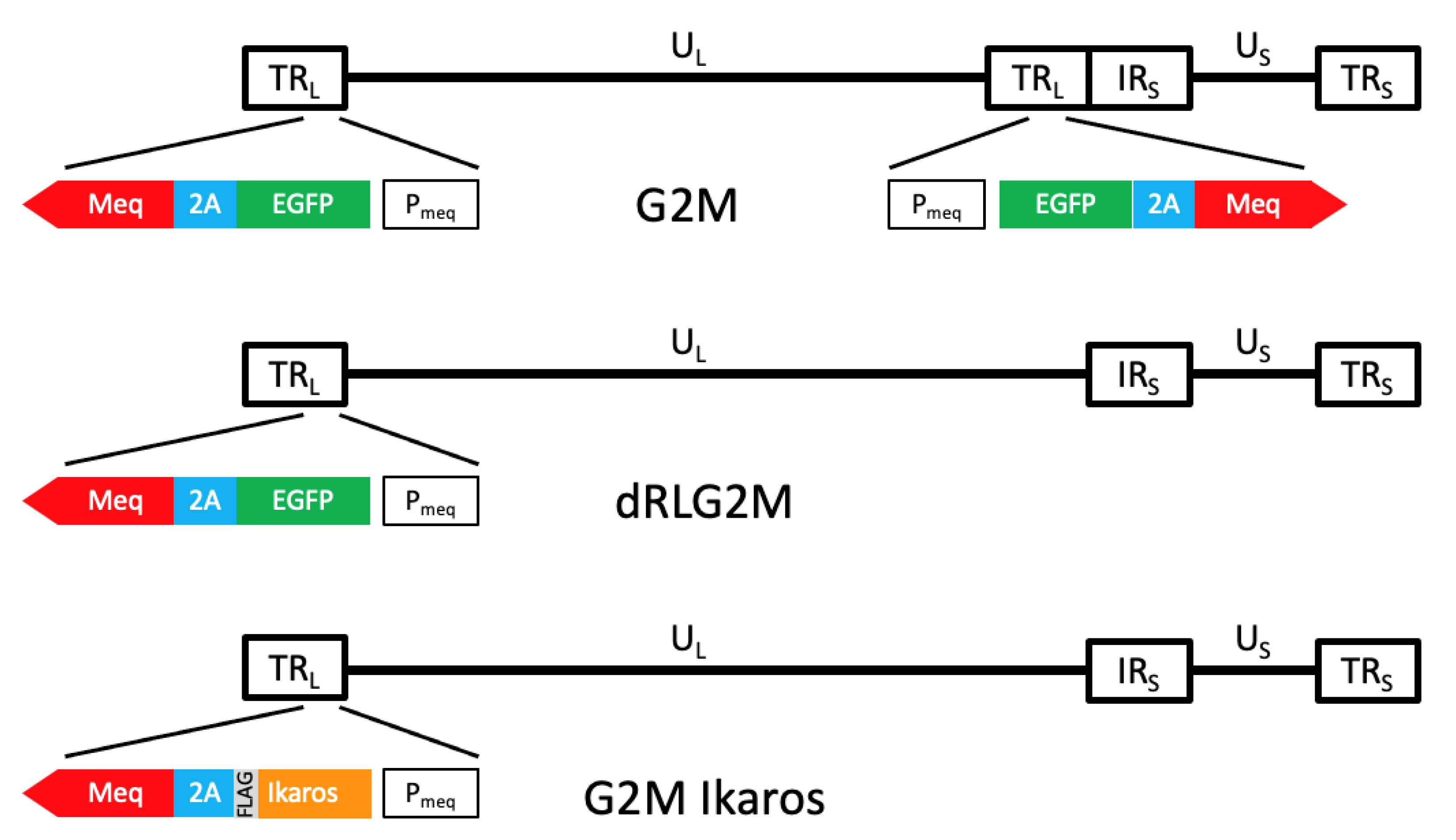
3.2. Validation That Mutant Ikaros Allele Promotes MDV-Induced Transformation
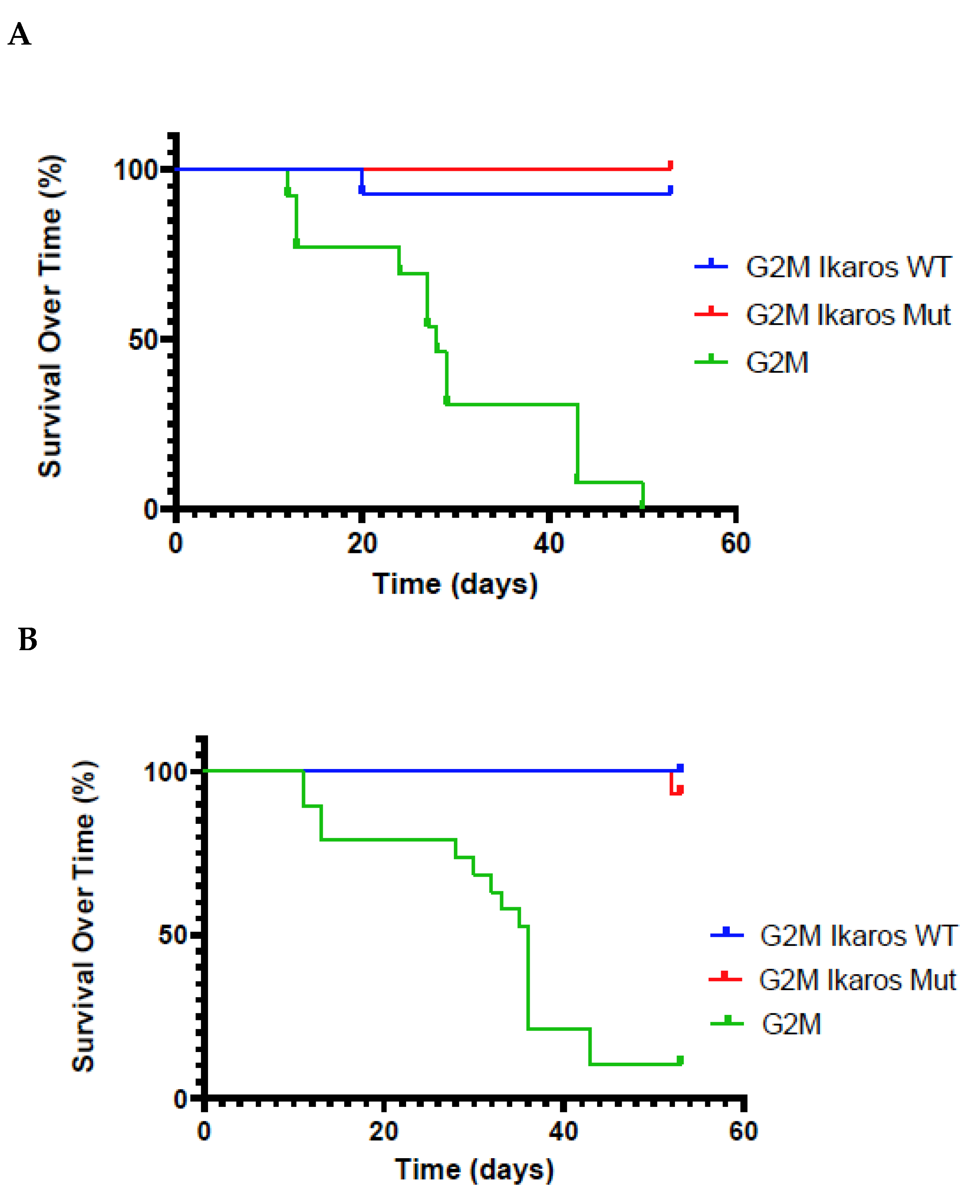
3.3. Lifespan of Birds in the G2M Treatment Group Were Significantly Shorter Than Other Groups
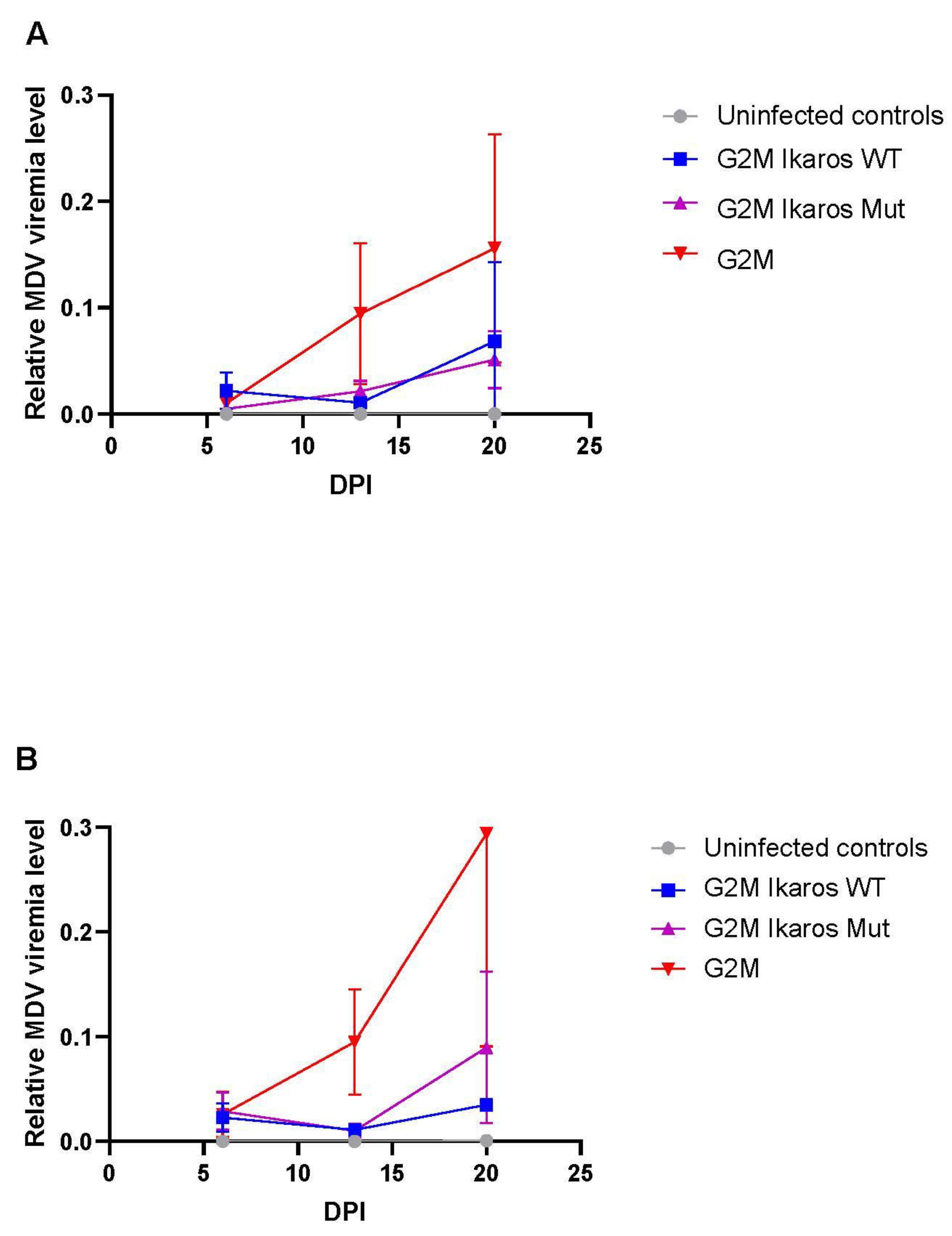
3.4. In Vivo Replication and Viremia of Recombinant MDVs Expressing Ikaros Alleles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD/FAO. Meat. In OECD-FAO Agricultural Outlook 2021-2030; OECD Publishing: Paris, France, 2021; pp. 163–177. [Google Scholar]
- Muir, W.M.; Wong, G.K.S.; Zhang, Y.; Wang, J.; Groenen, M.A.M.; Crooijmans, R.P.M.A.; Megens, H.J.; Zhang, H.; Okimoto, R.; Vereijken, A.; et al. Genome-wide assessment of worldwide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds. Proc. Natl. Acad. Sci. USA 2008, 105, 17312–17317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, C.; Fehler, F. Marek’s Disease: A Worldwide Problem. In Marek’s Disease, An Evolving Problem; Davison, F., Nair, V., Eds.; Elsevier Ltd.: Oxford, UK, 2004; pp. 49–61. [Google Scholar]
- Witter, R.L. Protective efficacy of Marek’s disease vaccines. Curr. Top. Micro. Immunol. 2001, 255, 57–90. [Google Scholar] [CrossRef]
- Witter, R.L. Increased virulence of Marek’s disease virus field isolates. Avian Dis. 1997, 41, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Davison, F.; Nair, V. Use of Marek’s disease vaccines: Could they be driving the virus to increasing virulence? Exp. Rev. Vacc. 2005, 4, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.E.; Read, A.F.; Walkden-Brown, S.; Savill, N.J.; Woolhouse, M.E.J. The effectiveness of mass vaccination on Marek’s disease virus (MDV) outbreaks and detection within a broiler barn: A modeling study. Epidemics 2013, 5, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect vaccination can enhance the transmission of highly virulent pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef] [PubMed]
- Kreager, K. Industry Concerns Workshop. In Current Research on Marek’s Disease; Silva, R.F., Cheng, H.H., Coussens, P.M., Lee, L., Velicer, L.F., Eds.; American Association of Avian Pathologists: Kennett Square, PA, USA, 1997; pp. 509–511. [Google Scholar]
- Reddy, S.M.; Izumiya, Y.; Lupiani, B. Marek’s disease vaccines: Current status, and strategies for improvement and development of vector vaccines. Vet. Microbiol. 2017, 206, 113–120. [Google Scholar] [CrossRef]
- Jones, D.; Lee, L.; Liu, J.L.; Kung, H.J.; Tillotson, J.K. Marek disease virus encodes a basic-leucine zipper gene resembling the Fos/Jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc. Natl. Acad. Sci. USA 1992, 89, 4042–4046. [Google Scholar] [CrossRef] [Green Version]
- Lupiani, B.; Lee, L.F.; Cui, X.; Gimeno, I.; Anderson, A.; Morgan, R.W.; Silva, R.F.; Witter, R.L.; Kung, H.J.; Reddy, S.M. Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 2004, 101, 11815–11820. [Google Scholar] [CrossRef] [Green Version]
- Conradie, A.M.; Bertzbach, L.D.; Trimpert, J.; Patria, J.N.; Murata, S.; Parcells, M.S.; Kaufer, B.B. Distinct polymorphisms in a single herpesvirus gene are capable of enhancing virulence and mediating vaccinal resistance. PLoS Pathog. 2020, 16, e1009104. [Google Scholar] [CrossRef]
- Trimpert, J.; Groenke, N.; Jenckel, M.; He, S.; Kunec, D.; Szpara, M.L.; Spatz, S.J.; Osterrieder, N.; McMahon, D.P. A phylogenomic analysis of Marek’s disease virus reveals independent paths to virulence in Eurasia and North America. Evol. Appl. 2017, 10, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.R.; Black Pyrkosz, A.; Steep, A.; Cheng, H.H. Identification of Marek’s disease virus genes associated with virulence of US strains. J. Gen. Virol. 2019, 100, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Delecluse, H.J.; Hammerschmidt, W. Status of Marek’s disease virus in established lymphoma cell lines: Herpesvirus integration is common. J. Virol. 1993, 67, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheimar, A.; Previdelli, R.L.; Wight, D.J.; Kaufer, B.B. Telomeres and telomerase: Role in Marek’s disease virus pathogenesis, integration and tumorigenesis. Viruses 2017, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; Hunt, H.D.; Cheng, H.; Delany, M. Chromosomal integration of an avian oncogenic herpesvirus reveals telomeric preferences and evidence for lymphoma clonality. Herpesviridae 2010, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.M.; Cheng, H.H.; Delany, M.E. Temporal kinetics of Marek’s disease herpesvirus: Integration occurs early after infection in both B and T Cells. Cytogen. Genome Res. 2014, 144, 142–154. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Stephens, E.A.; Witter, R.L.; Lee, L.F.; Sharma, J.M.; Nazerian, K.; Longenecker, B.M. Characteristics of JMV Marek’s disease tumor: A nonproductively infected transplantable cell lacking in rescuable virus. J. Natl. Cancer Inst. 1976, 57, 865–874. [Google Scholar] [CrossRef]
- Tai, S.S.; Hearn, C.; Umthong, S.; Agafitei, O.; Cheng, H.H.; Dunn, J.R.; Niikura, M. Expression of Marek’s disease virus oncoprotein Meq during infection in the natural host. Virology 2017, 503, 103–113. [Google Scholar] [CrossRef]
- Engel, A.T.; Selvaraj, R.K.; Kamil, J.P.; Osterrieder, N.; Kaufer, B.B. Marek’s disease viral interleukin-8 promotes lymphoma formation through targeted recruitment of B cells and CD4+ CD25+ T cells. J. Virol. 2012, 86, 8536–8545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef]
- Hildebrandt, E.; Dunn, J.R.; Perumbakkam, S.; Niikura, M.; Cheng, H.H. Characterizing the molecular basis of attenuation of Marek’s disease virus via in vitro serial passage identifies de novo mutations in the helicase-primase subunit gene UL5 and other candidates associated with reduced virulence. J. Virol. 2014, 88, 6232–6342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.R.; Silva, R.F. Ability of MEQ-deleted MDV vaccine candidates to adversely affect lymphoid organs and chicken weight gain. Avian Dis. 2012, 56, 494–500. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Proto. Bioinfo. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Andrews, S.; Babraham, I. FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 May 2014).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2013, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broad Institute. Available online: http://broadinstitute.github.io/picard/ (accessed on 22 February 2016).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Xi, L.; Hughes, D.S.; Zhang, J.; Zhang, J.; Futreal, P.A.; Wheeler, D.A.; Wang, W. MuSE: Accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016, 17, 178. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.; Carter, S.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotech. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Roth, A.; Ding, J.; Morin, R.; Crisan, A.; Ha, G.; Giuliany, R.; Bashashati, A.; Hirst, M.; Turashvili, G.; Oloumi, A.; et al. JointSNVMix: A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012, 28, 907–913. [Google Scholar] [CrossRef]
- Larson, D.; Harris, C.; Chen, K.; Koboldt, D.; Abbott, T.; Dooling, D.; Ley, T.; Mardis, E.; Wilson, R.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, C.; Dry, J. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucl. Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Koboldt, D.; Zhang, Q.; Larson, D.; Shen, D.; McLellan, M.; Lin, L.; Miller, C.; Mardis, E.; Ding, L.; Wilson, R. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 5, D635–D642. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 1993, 9, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.H.; Perumbakkam, S.; Black Pyrkosz, A.; Dunn, J.R.; Legarra, A.; Muir, W.M. Fine mapping of QTL and genomic prediction using allele-specific expression SNPs demonstrates that the complex trait of genetic resistance to Marek’s disease is predominantly determined by transcriptional regulation. BMC Genomics 2015, 16, 816. [Google Scholar] [CrossRef] [Green Version]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef]
- Harker, N.; Naito, T.; Cortes, M.; Hostert, A.; Hirschberg, S.; Tolaini, M.; Roderick, K.; Georgopoulos, K.; Kioussis, D. The CD8-alpha gene locus is regulated by the Ikaros family of proteins. Molec. Cell 2002, 10, 1403–1415. [Google Scholar] [CrossRef]
- Powell, M.D.; Read, K.A.; Sreekumar, B.K.; Oestreich, K.J. Ikaros zinc finger transcription factors: Regulators of cytokine signaling pathways and CD4+ T helper cell differentiation. Front. Immunol. 2019, 10, 1299. [Google Scholar] [CrossRef] [Green Version]
- Haire, R.N.; Miracle, A.L.; Rast, J.P.; Litman, G.W. Members of the Ikaros gene family are present in early representative vertebrates. J. Immunol. 2000, 165, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Nera, K.P.; Alinikula, J.; Terho, P.; Narvi, E.; Törnquist, K.; Kurosaki, T.; Buerstedde, J.M.; Lassila, O. Ikaros has a crucial role in regulation of B cell receptor signaling. Eur. J. Immunol. 2006, 36, 516–525. [Google Scholar] [CrossRef]
- Yoshia, T.; Georgopoulos, K. Ikaros fingers on lymphocyte differentiation. Int. J. Hematol. 2014, 100, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Winandy, S.; Wu, P.; Georgopoulos, K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell 1995, 83, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, P.; Perkins, A.C.; Cobb, B.S.; Ferrini, R.; Sridharan, R.; Hoyne, G.F.; Nelms, K.A.; Smale, S.T.; Goodnow, C.C. Widespread failure of hematolymphoid differentiation caused by a recessive niche-filling allele of the Ikaros transcription factor. Immunity 2003, 19, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jackson, A.F.; Naito, T.; Dose, M.; Seavitt, J.; Liu, F.; Heller, E.J.; Kashiwagi, M.; Yoshida, T.; Gounari, F.; et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol. 2011, 13, 86–94. [Google Scholar] [CrossRef]
- Schwickert, T.A.; Tagoh, H.; Gültekin, S.; Dakic, A.; Axelsson, E.; Minnich, M.; Ebert, A.; Werner, B.; Roth, M.; Cimmino, L.; et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat. Immunol. 2014, 15, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.; Downing, J. Ikaros and acute leukemia. Leuk. Lymphoma 2008, 49, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.D.; Strassburger, P.; Du Pasquier, L. Conservation of a master hematopoietic switch gene during vertebrate evolution: Isolation and characterization of Ikaros from teleost and amphibian species. Eur. J. Immunol. 1997, 27, 3049–3058. [Google Scholar] [CrossRef]
- Payne, M.A. Zinc finger structure-function in Ikaros. World J. Biol. Chem. 2011, 2, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Dijon, M.; Bardin, F.; Murati, A.; Batoz, M.; Chabannon, C.; Tonnelle, C. The role of Ikaros in human erythroid differentiation. Blood 2008, 111, 1138–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, M.; Zhang, H.; Cao, S.; Li, Y.; Jiang, S.; Song, Y.; Liu, S. Detection of p53 mutation and serum monitoring alert caused by Marek’s disease virus in poultry. BMC Vet. Res. 2020, 16, 303. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.M.; Izumiya, Y.; Brunovskis, P.; Xia, L.; Parcells, M.S.; Reddy, S.M.; Lee, L.; Chen, H.W.; Kung, H.J. Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek’s disease virus-transformed T cells. J. Virol. 2003, 77, 12841–12851. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Ye, Y.; Lee, L.F.; Kung, H.J. Transforming potential of the herpesvirus oncoprotein MEQ: Morphological transformation, serum-independent growth, and inhibition of apoptosis. J. Virol. 1998, 72, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Li, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Jin, Y.; Chen, H.; Ding, C.; Li, L.; et al. The Meq oncoprotein of Marek’s disease virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Virol, J. 2010, 7, 348. [Google Scholar] [CrossRef] [Green Version]
- Iempridee, T.; Reusch, J.A.; Riching, A.; Johannsen, E.C.; Dovat, S.; Kenney, S.C.; Mertza, J.E. Epstein-Barr virus utilizes Ikaros in regulating its latent-lytic switch in B cells. J. Virol. 2014, 88, 4811–4827. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position 1 | Reference 2 | Alternative 3 | Variant Type | AA Change | Sample 4 | Prediction |
|---|---|---|---|---|---|---|
| 80,972,101 | C | T | missense | Arg162Cys | 777, 851, 901 | deleterious |
| 80,972,102 | G | T | missense | Arg162Leu | 835 | deleterious |
| 80,972,104 | C | T | missense | His163Tyr | 842, 927 | deleterious |
| 80,972,116 | C | T | missense | His167Tyr | 901 | deleterious |
| 80,972,118 | C | G | missense | His167Gln | 756 | deleterious |
| 80,972,141 | G | A | missense | Cys175Tyr | 901 | deleterious |
| 80,972,141 | G | GCCA | inframe insertion | His176dup | 901 | deleterious |
| 80,972,149 | TGTAACTACGCCTGCCGGCGCA | T | inframe deletion | Cys178 to Arg185 delinsTrp | 911 | deleterious |
| 80,972,152 | A | AACT | inframe insertion | Tyr180dup | 918 | deleterious |
| 80,972,167 | C | T | missense | Arg184Cys | 927 | deleterious |
| Replicate | Treatment | Total Birds | MD 1 | Tumor Positive | ||
|---|---|---|---|---|---|---|
| Count | Percent | Count | Percent | |||
| 1 | none | 6 | 0 | 0 | 0 | 0 |
| G2M | 13 | 4 | 31 | 2 | 15 | |
| G2M Ikaros WT | 14 | 2 | 14 | 2 | 14 | |
| G2M Ikaros Mut | 14 | 13 | 93 | 12 | 86 | |
| 2 | none | 10 | 0 | 0 | 0 | 0 |
| G2M | 19 | 9 | 47 | 7 | 37 | |
| G2M Ikaros WT | 19 | 2 | 11 | 2 | 11 | |
| G2M Ikaros Mut | 15 | 14 | 93 | 14 | 93 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steep, A.; Hildebrandt, E.; Xu, H.; Hearn, C.; Frishman, D.; Niikura, M.; Dunn, J.R.; Kim, T.; Conrad, S.J.; Muir, W.M.; et al. Identification and Validation of Ikaros (IKZF1) as a Cancer Driver Gene for Marek’s Disease Virus-Induced Lymphomas. Microorganisms 2022, 10, 401. https://doi.org/10.3390/microorganisms10020401
Steep A, Hildebrandt E, Xu H, Hearn C, Frishman D, Niikura M, Dunn JR, Kim T, Conrad SJ, Muir WM, et al. Identification and Validation of Ikaros (IKZF1) as a Cancer Driver Gene for Marek’s Disease Virus-Induced Lymphomas. Microorganisms. 2022; 10(2):401. https://doi.org/10.3390/microorganisms10020401
Chicago/Turabian StyleSteep, Alec, Evin Hildebrandt, Hongen Xu, Cari Hearn, Dmitrij Frishman, Masahiro Niikura, John R. Dunn, Taejoong Kim, Steven J. Conrad, William M. Muir, and et al. 2022. "Identification and Validation of Ikaros (IKZF1) as a Cancer Driver Gene for Marek’s Disease Virus-Induced Lymphomas" Microorganisms 10, no. 2: 401. https://doi.org/10.3390/microorganisms10020401