
Mastadenovirus Molecular Diversity in Waste and Environmental Waters from the Lisbon Metropolitan Area
 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
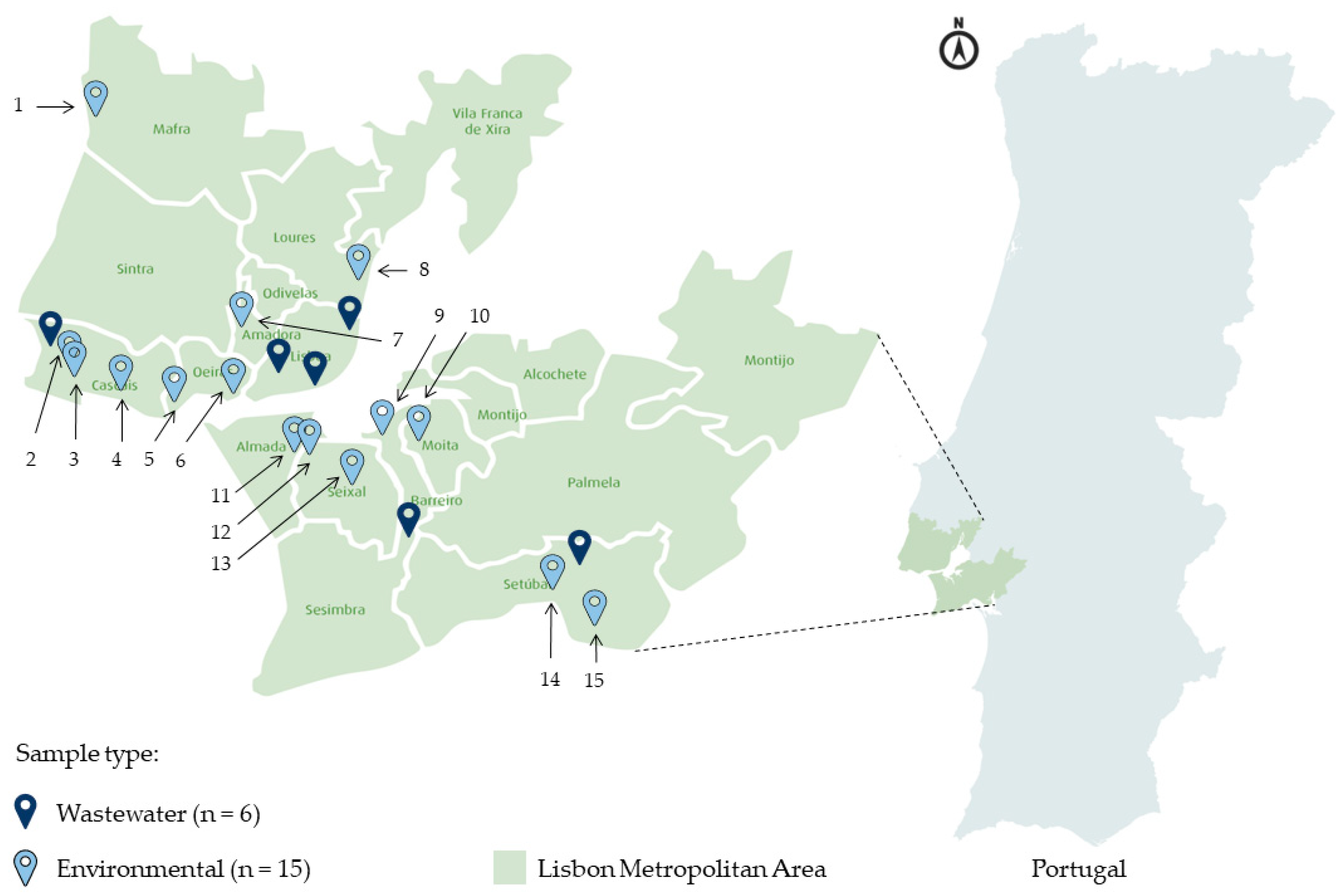
2.1. Sample Collection
2.2. Viral Particles Concentration by Skimmed Milk Flocculation, and DNA Extraction
2.3. Primers Design
2.4. Nested Touch-Down PCR Assays
2.5. DNA Cloning and Sanger Sequencing
2.6. NGS Sequencing
2.7. Nucleotide Sequences Analysis
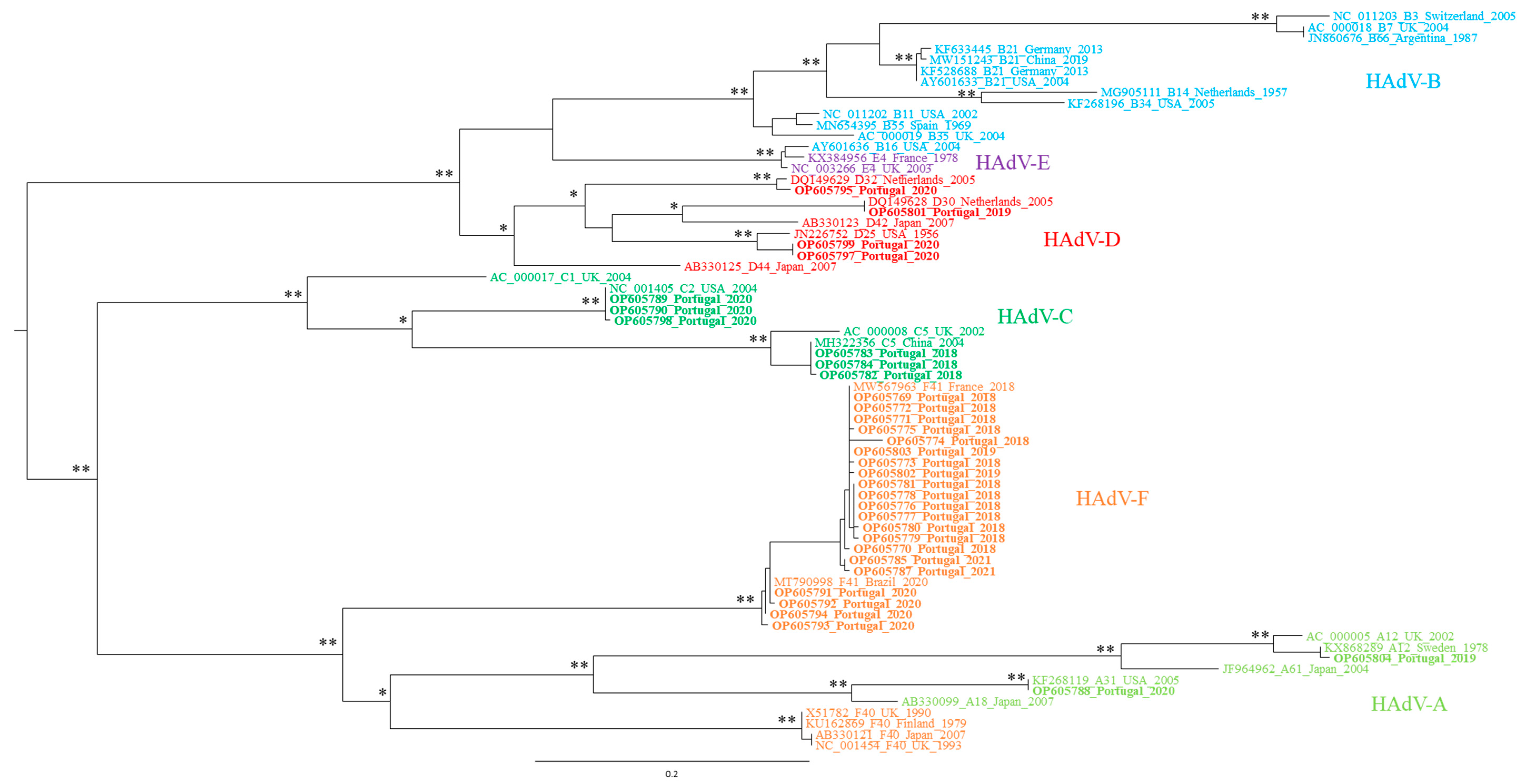
2.8. Phylogenetic Analysis
3. Results
3.1. HAdV Screening in the Collected Water Samples
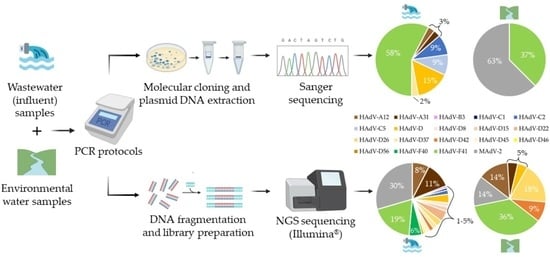
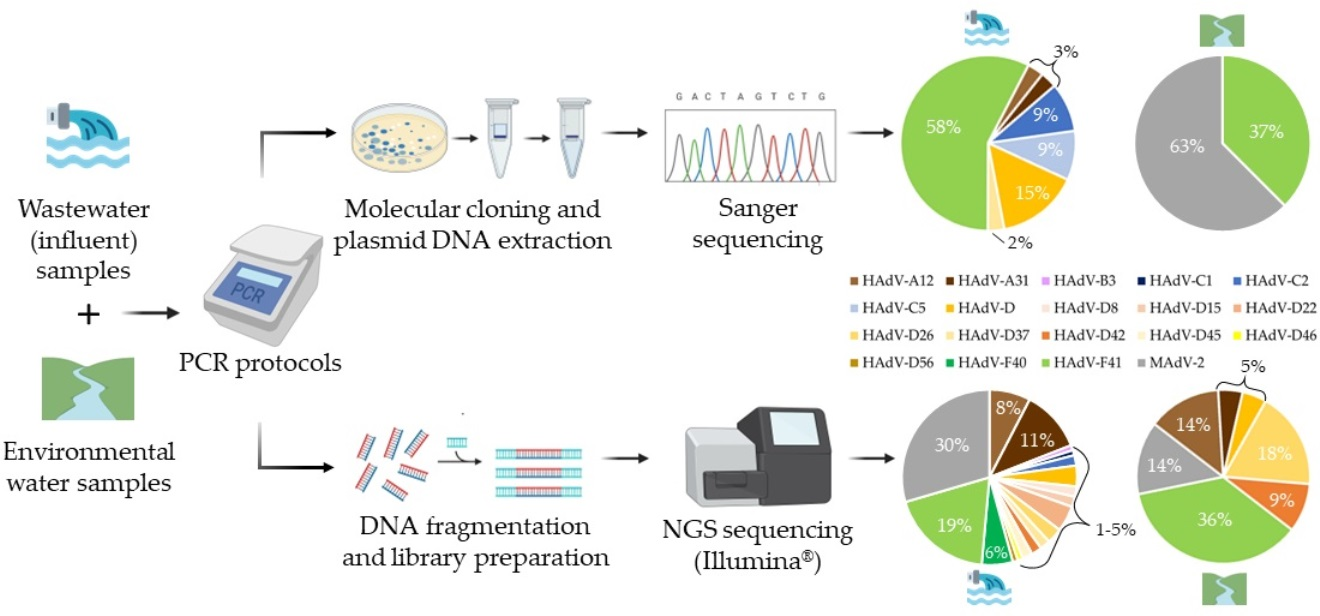
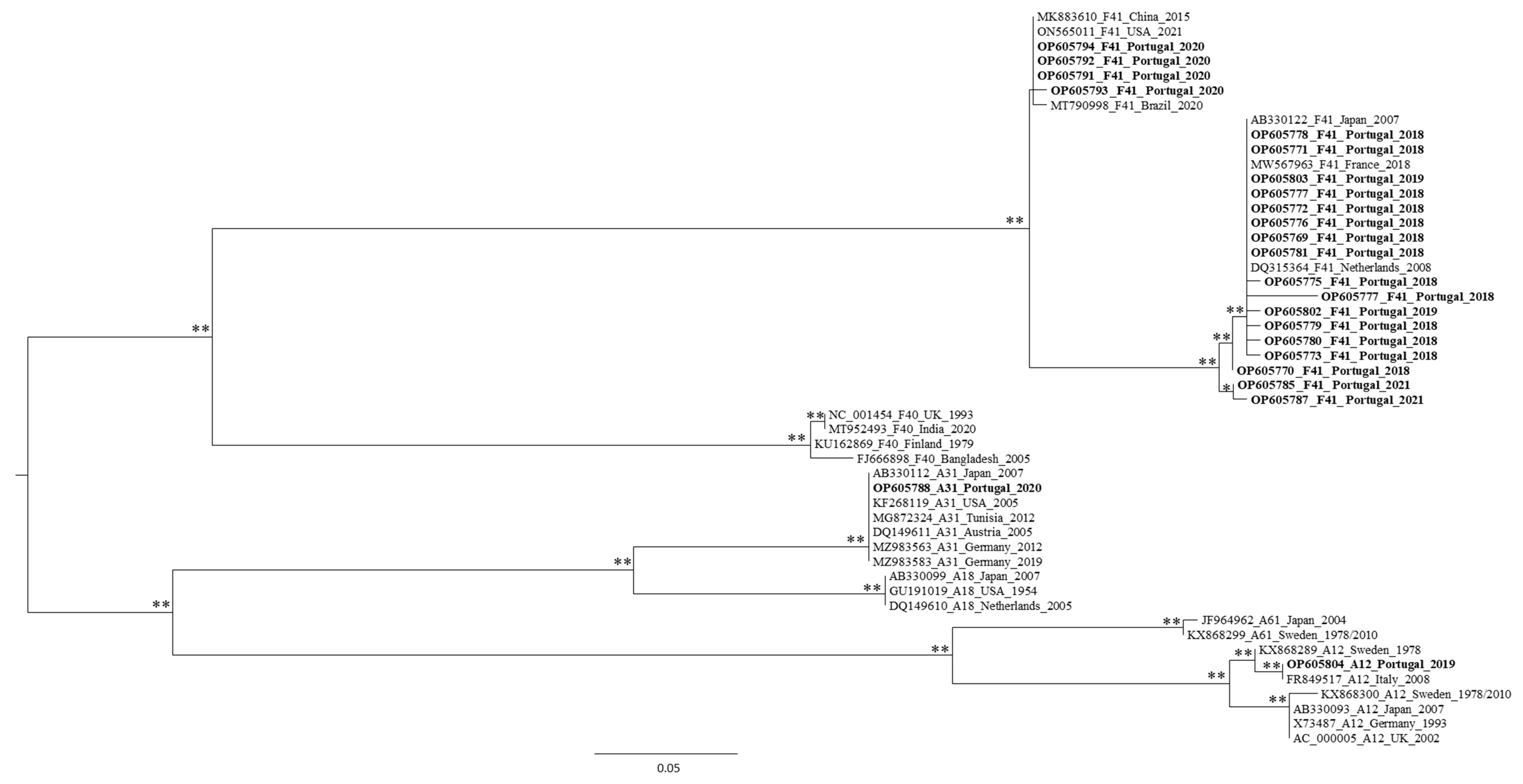
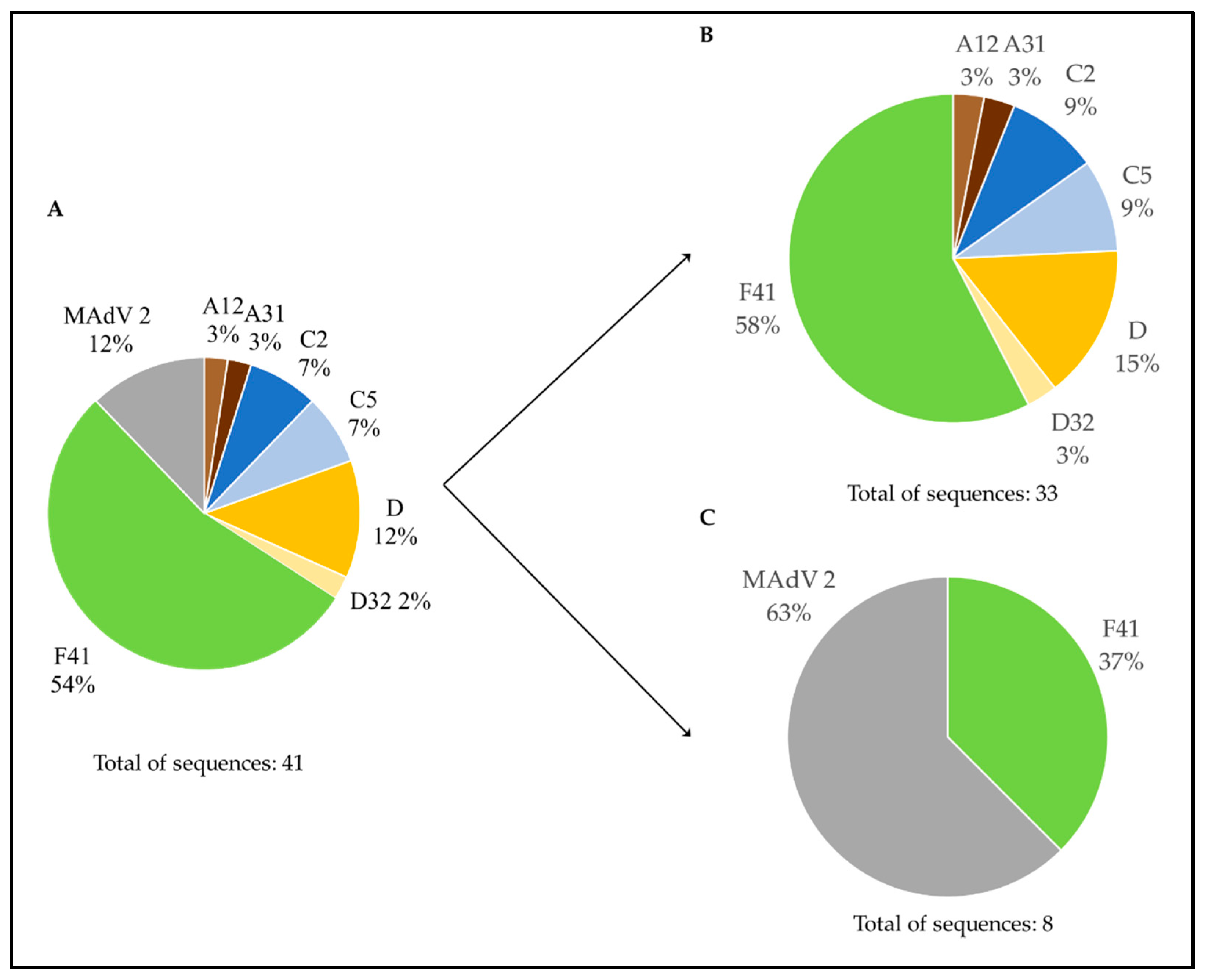
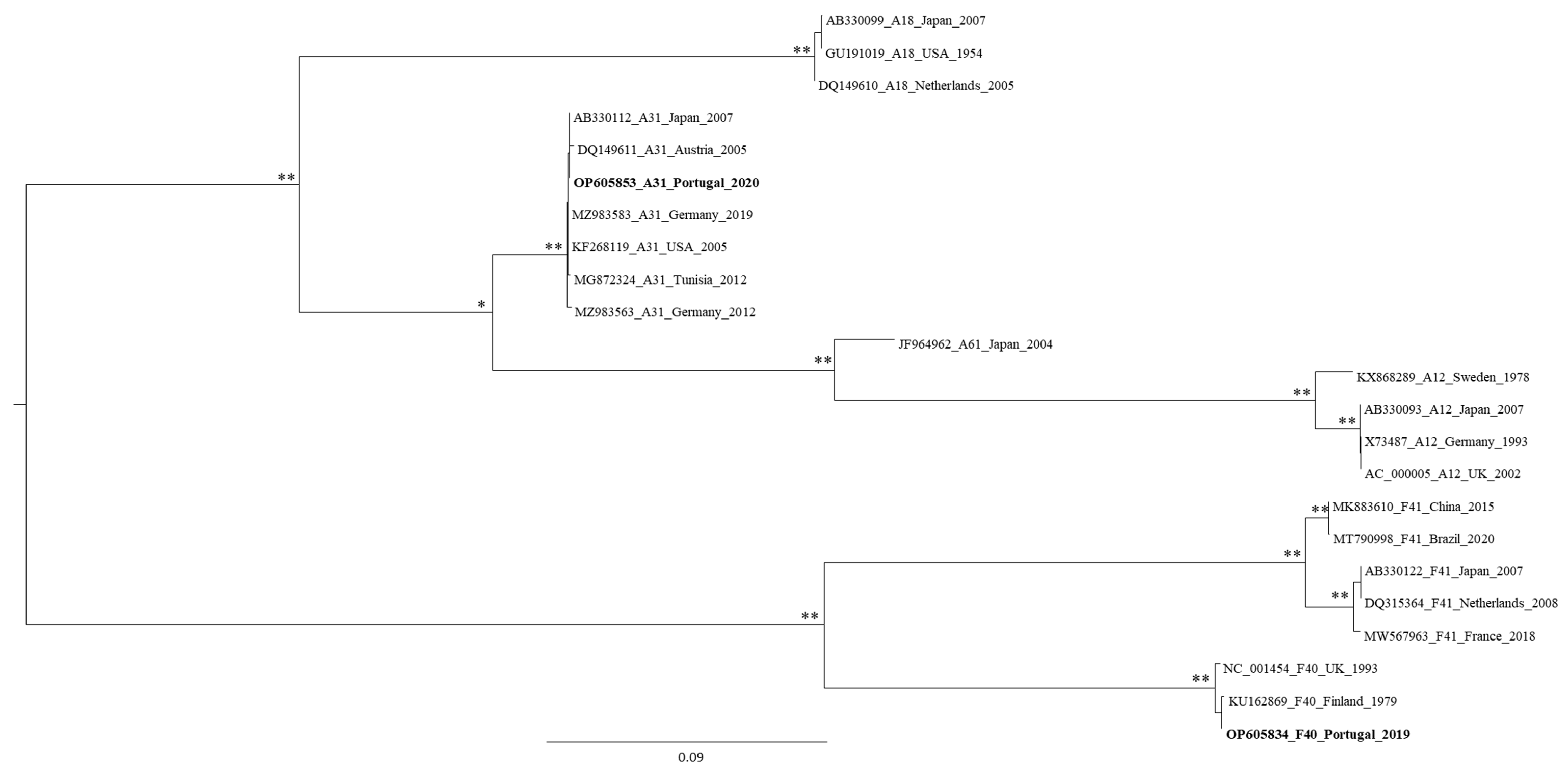
3.2. Genetic Characterization of AdV, as Defined by Molecular Cloning Followed by Sanger Sequencing
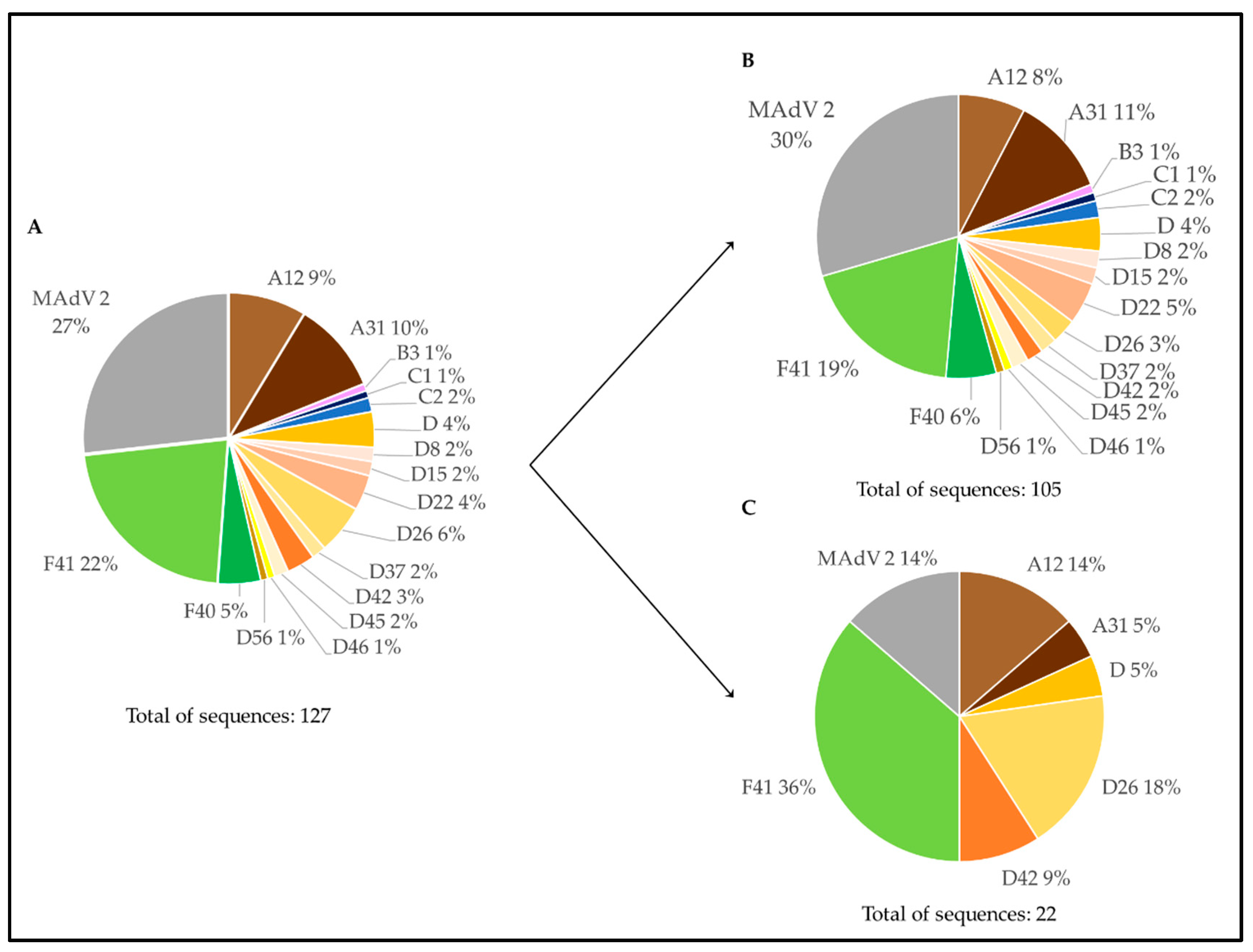
3.3. Genetic Characterization of AdV Sequences as Defined by the Analysis of AdV Amplicons Using an NGS (Illumina) Approach
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verani, M.; Federigi, I.; Donzelli, G.; Cioni, L.; Carducci, A. Human Adenoviruses as Waterborne Index Pathogens and Their Use for Quantitative Microbial Risk Assessment. Sci. Total Environ. 2019, 651, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Calgua, B.; Fumian, T.; Rusiñol, M.; Rodriguez-Manzano, J.; Mbayed, V.A.; Bofill-Mas, S.; Miagostovich, M.; Girones, R. Detection and Quantification of Classic and Emerging Viruses by Skimmed-Milk Flocculation and PCR in River Water from Two Geographical Areas. Water Res. 2013, 47, 2797–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, T.-T.; Lipp, E.K. Enteric Viruses of Humans and Animals in Aquatic Environments: Health Risks, Detection, and Potential Water Quality Assessment Tools. Microbiol. Mol. Biol. Rev. 2005, 69, 357–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, P.; Suttie, M.; Lewis, V.T.; Smith, A.P.; Singer, A.C. Detection of Untreated Sewage Discharges to Watercourses Using Machine Learning. Npj Clean Water 2021, 4, 18. [Google Scholar] [CrossRef]
- Sato, T.; Qadir, M.; Yamamoto, S.; Endo, T.; Zahoor, A. Global, Regional, and Country Level Need for Data on Wastewater Generation, Treatment, and Use. Agric. Water Manag. 2013, 130, 1–13. [Google Scholar] [CrossRef]
- Ullah Bhat, S.; Qayoom, U. Implications of Sewage Discharge on Freshwater Ecosystems. In Sewage-Recent Advances, New Perspectives and Applications; Zhang, T., Ed.; IntechOpen: London, UK, 2022; ISBN 978-1-83969-824-8. [Google Scholar]
- Almeida, P.; Albuquerque, T.; Antunes, M.; Ferreira, A.; Pelletier, G. Effects of Wastewater Treatment Plant’s Discharges on a Freshwater Ecosystem—A Case Study on the Ramalhoso River (Portugal). Water Air Soil Pollut. 2021, 232, 206. [Google Scholar] [CrossRef]
- Opere, W.M.; John, M.; Ombori, O. Occurrence of Enteric Viruses in Surface Water and the Relationship with Changes in Season and Physical Water Quality Dynamics. Adv. Virol. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- La Rosa, G.; Pourshaban, M.; Iaconelli, M.; Muscillo, M. Quantitative Real-Time PCR of Enteric Viruses in Influent and Effluent Samples from Wastewater Treatment Plants in Italy. Ann. Ist Super. SanItà 2010, 46, 266–273. [Google Scholar] [CrossRef]
- Kokkinos, P.A.; Ziros, P.G.; Mpalasopoulou, A.; Galanis, A.; Vantarakis, A. Molecular Detection of Multiple Viral Targets in Untreated Urban Sewage from Greece. Virol. J. 2011, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Gonzales-Gustavson, E.; Rusiñol, M.; Medema, G.; Calvo, M.; Girones, R. Quantitative Risk Assessment of Norovirus and Adenovirus for the Use of Reclaimed Water to Irrigate Lettuce in Catalonia. Water Res. 2019, 153, 91–99. [Google Scholar] [CrossRef]
- Iaconelli, M.; Muscillo, M.; Della Libera, S.; Fratini, M.; Meucci, L.; De Ceglia, M.; Giacosa, D.; La Rosa, G. One-Year Surveillance of Human Enteric Viruses in Raw and Treated Wastewaters, Downstream River Waters, and Drinking Waters. Food Environ. Virol. 2017, 9, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.; Greening, G.E.; Leonard, M.; Lewis, G.D. Evaluation of Human Adenovirus and Human Polyomavirus as Indicators of Human Sewage Contamination in the Aquatic Environment. Water Res. 2013, 47, 6750–6761. [Google Scholar] [CrossRef] [PubMed]
- Wyn-Jones, A.P.; Carducci, A.; Cook, N.; D’Agostino, M.; Divizia, M.; Fleischer, J.; Gantzer, C.; Gawler, A.; Girones, R.; Höller, C.; et al. Surveillance of Adenoviruses and Noroviruses in European Recreational Waters. Water Res. 2011, 45, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Bofill-Mas, S.; Albinana-Gimenez, N.; Clemente-Casares, P.; Hundesa, A.; Rodriguez-Manzano, J.; Allard, A.; Calvo, M.; Girones, R. Quantification and Stability of Human Adenoviruses and Polyomavirus JCPyV in Wastewater Matrices. Appl. Environ. Microbiol. 2006, 72, 7894–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rames, E.; Roiko, A.; Stratton, H.; Macdonald, J. Technical Aspects of Using Human Adenovirus as a Viral Water Quality Indicator. Water Res. 2016, 96, 308–326. [Google Scholar] [CrossRef] [PubMed]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kaján, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae 2022: This Article Is Part of the ICTV Virus Taxonomy Profiles Collection. J. Gen. Virol. 2022, 103, 1721. [Google Scholar] [CrossRef] [PubMed]
- Fenner’s Veterinary Virology, 5th ed.; Maclachlan, N.J.; Dubovi, E.J.; Barthold, S.W.; Swayne, D.E.; Winton, J.R. (Eds.) Elsevier/AP, Academic Press is an imprint of Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-800946-8. [Google Scholar]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A Review of 65 Years of Human Adenovirus Seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef]
- Gallardo, J.; Pérez-Illana, M.; Martín-González, N.; San Martín, C. Adenovirus Structure: What Is New? Int. J. Mol. Sci. 2021, 22, 5240. [Google Scholar] [CrossRef]
- Besson, S.; Vragniau, C.; Vassal-Stermann, E.; Dagher, M.C.; Fender, P. The Adenovirus Dodecahedron: Beyond the Platonic Story. Viruses 2020, 12, 718. [Google Scholar] [CrossRef]
- Crenshaw, B.J.; Jones, L.B.; Bell, C.R.; Kumar, S.; Matthews, Q.L. Perspective on Adenoviruses: Epidemiology, Pathogenicity, and Gene Therapy. Biomedicines 2019, 7, 61. [Google Scholar] [CrossRef]
- Lynch, J.; Kajon, A. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaconelli, M.; Valdazo-González, B.; Equestre, M.; Ciccaglione, A.R.; Marcantonio, C.; Della Libera, S.; La Rosa, G. Molecular Characterization of Human Adenoviruses in Urban Wastewaters Using next Generation and Sanger Sequencing. Water Res. 2017, 121, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Allard, A.; Vantarakis, A. Adenoviruses. In Water and Sanitation for the 21st Century: Health and Microbiological Aspects of Excreta and Wastewater Management (Global Water Pathogen Project); Michigan State University, Rose, J.B., Jiménez Cisneros, B., UNESCO-International Hydrological Programme, Eds.; Michigan State University: East Lansing, MI, USA, 2017; ISBN 978-0-9967252-8-6. [Google Scholar]
- Lun, J.H.; Crosbie, N.D.; White, P.A. Genetic Diversity and Quantification of Human Mastadenoviruses in Wastewater from Sydney and Melbourne, Australia. Sci. Total Environ. 2019, 675, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Cook, J.L. Human Adenovirus Infections: Update and Consideration of Mechanisms of Viral Persistence. Curr. Opin. Infect. Dis. 2018, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Diagnosis, Treatment and Prevention of Virus Infections. In Encyclopedia of Virology; Heim, A. (Ed.) Academic Press, Elsevier: Amsterdam, The Netherlands; Boston, MA, USA; Heidelberg, Germany, 2021; Volume 5, pp. 197–205. ISBN 978-0-12-814516-6. [Google Scholar]
- Sedji, M.I.; Varbanov, M.; Meo, M.; Colin, M.; Mathieu, L.; Bertrand, I. Quantification of Human Adenovirus and Norovirus in River Water in the North-East of France. Environ. Sci. Pollut. Res. 2018, 25, 30497–30507. [Google Scholar] [CrossRef]
- Mena, K.D.; Gerba, C.P. Waterborne Adenovirus. In Reviews of Environmental Contamination and Toxicology; Reviews of Environmental Contamination and Toxicology; Springer: New York, NY, USA, 2009; Volume 198, pp. 1–35. ISBN 978-0-387-09646-9. [Google Scholar]
- Lee, S.-H.; Kim, S.-J. Detection of Infectious Enteroviruses and Adenoviruses in Tap Water in Urban Areas in Korea. Water Res. 2002, 36, 248–256. [Google Scholar] [CrossRef]
- Bisseux, M.; Colombet, J.; Mirand, A.; Roque-Afonso, A.-M.; Abravanel, F.; Izopet, J.; Archimbaud, C.; Peigue-Lafeuille, H.; Debroas, D.; Bailly, J.-L.; et al. Monitoring Human Enteric Viruses in Wastewater and Relevance to Infections Encountered in the Clinical Setting: A One-Year Experiment in Central France, 2014 to 2015. Eurosurveillance 2018, 23, 17-00237. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, G.; Sanseverino, I.; Della Libera, S.; Iaconelli, M.; Ferrero, V.E.V.; Barra Caracciolo, A.; Lettieri, T. The Impact of Anthropogenic Pressure on the Virological Quality of Water from the Tiber River, Italy. Lett. Appl. Microbiol. 2017, 65, 298–305. [Google Scholar] [CrossRef]
- Rusiñol, M.; Fernandez-Cassi, X.; Timoneda, N.; Carratalà, A.; Abril, J.F.; Silvera, C.; Figueras, M.J.; Gelati, E.; Rodó, X.; Kay, D.; et al. Evidence of Viral Dissemination and Seasonality in a Mediterranean River Catchment: Implications for Water Pollution Management. J. Environ. Manag. 2015, 159, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Grøndahl-Rosado, R.C.; Yarovitsyna, E.; Trettenes, E.; Myrmel, M.; Robertson, L.J. A One Year Study on the Concentrations of Norovirus and Enteric Adenoviruses in Wastewater and A Surface Drinking Water Source in Norway. Food Environ. Virol. 2014, 6, 232–245. [Google Scholar] [CrossRef]
- Maunula, L.; Söderberg, K.; Vahtera, H.; Vuorilehto, V.-P.; von Bonsdorff, C.-H.; Valtari, M.; Laakso, T.; Lahti, K. Presence of Human Noro- and Adenoviruses in River and Treated Wastewater, a Longitudinal Study and Method Comparison. J. Water Health 2012, 10, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuolale, O.; Okoh, A. Incidence of Human Adenoviruses and Hepatitis A Virus in the Final Effluent of Selected Wastewater Treatment Plants in Eastern Cape Province, South Africa. Virol. J. 2015, 12, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staggemeier, R.; Heck, T.M.S.; Demoliner, M.; Ritzel, R.G.F.; Röhnelt, N.M.S.; Girardi, V.; Venker, C.A.; Spilki, F.R. Enteric Viruses and Adenovirus Diversity in Waters from 2016 Olympic Venues. Sci. Total Environ. 2017, 586, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Quintão, T.S.C.; Silva, F.G.; Pereira, A.L.; Araújo, W.N.; Oliveira, P.M.; Souza, M.B.L.D.; Lamounier, T.A.; Haddad, R. Detection and Molecular Characterization of Enteric Adenovirus in Treated Wastewater in the Brazilian Federal District. SN Appl. Sci. 2021, 3, 691. [Google Scholar] [CrossRef]
- Artieda, J.; Piñeiro, L.; González, M.C.; Muñoz, M.J.; Basterrechea, M.; Iturzaeta, A.; Cilla, G. A Swimming Pool-Related Outbreak of Pharyngoconjunctival Fever in Children Due to Adenovirus Type 4, Gipuzkoa, Spain, 2008. Eurosurveillance 2009, 14, 19125. [Google Scholar] [CrossRef]
- Farkas, K.; Cooper, D.M.; McDonald, J.E.; Malham, S.K.; de Rougemont, A.; Jones, D.L. Seasonal and Spatial Dynamics of Enteric Viruses in Wastewater and in Riverine and Estuarine Receiving Waters. Sci. Total Environ. 2018, 634, 1174–1183. [Google Scholar] [CrossRef]
- Rebelo-de-Andrade, H.; Pereira, C.; Gíria, M.; Prudêncio, E.; Brito, M.J.; Calé, E.; Taveira, N. Outbreak of Acute Respiratory Infection among Infants in Lisbon, Portugal, Caused by Human Adenovirus Serotype 3 and a New 7/3 Recombinant Strain. J. Clin. Microbiol. 2010, 48, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, G.; Gouveia, E.; Mesquita, J.R.; Almeida, A.; Ribeiro, A.; Rocha-Pereira, J.; São José Nascimento, M. Outbreak of Acute Gastroenteritis Caused by Adenovirus Type 41 in a Kindergarten. Epidemiol. Infect. 2011, 139, 1672–1675. [Google Scholar] [CrossRef]
- Ribeiro, J.; Ferreira, D.; Arrabalde, C.; Almeida, S.; Baldaque, I.; Sousa, H. Prevalence of Adenovirus and Rotavirus Infection in Immunocompromised Patients with Acute Gastroenteritis in Portugal. World J. Virol. 2015, 4, 372. [Google Scholar] [CrossRef]
- Ribeiro, A.; Ramalheira, E.; Cunha, Â.; Gomes, N.C.M.; Almeida, A. Incidence of Rotavirus and Adenovirus: Detection by Molecular and Immunological Methods in Human Faeces. J. Pure Appl. Microbiol. 2013, 7, 1505–1513. [Google Scholar]
- Hjelmsø, M.H.; Hellmér, M.; Fernandez-Cassi, X.; Timoneda, N.; Lukjancenko, O.; Seidel, M.; Elsässer, D.; Aarestrup, F.M.; Löfström, C.; Bofill-Mas, S.; et al. Evaluation of Methods for the Concentration and Extraction of Viruses from Sewage in the Context of Metagenomic Sequencing. PLoS ONE 2017, 12, e0170199. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Leitner, T. PrimerDesign-M: A Multiple-Alignment Based Multiple-Primer Design Tool for Walking across Variable Genomes. Bioinformatics 2015, 31, 1472–1474. [Google Scholar] [CrossRef] [Green Version]
- Avellón, A.; Pérez, P.; Aguilar, J.C.; ortiz de Lejarazu, R.; Echevarría, J.E. Rapid and Sensitive Diagnosis of Human Adenovirus Infections by a Generic Polymerase Chain Reaction. J. Virol. Methods 2001, 92, 113–120. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Filipa-Silva, A.; Parreira, R.; Martínez-Puchol, S.; Bofill-Mas, S.; Barreto Crespo, M.T.; Nunes, M. The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology. Foods 2020, 9, 1634. [Google Scholar] [CrossRef]
- Ibrahim, C.; Hassen, A.; Pothier, P.; Mejri, S.; Hammami, S. Molecular Detection and Genotypic Characterization of Enteric Adenoviruses in a Hospital Wastewater. Environ. Sci. Pollut. Res. 2018, 25, 10977–10987. [Google Scholar] [CrossRef]
- Nour, I.; Hanif, A.; Zakri, A.M.; Al-Ashkar, I.; Alhetheel, A.; Eifan, S. Human Adenovirus Molecular Characterization in Various Water Environments and Seasonal Impacts in Riyadh, Saudi Arabia. Int. J. Environ. Res. Public Health 2021, 18, 4773. [Google Scholar] [CrossRef]
- Monteiro, S.; Ebdon, J.; Santos, R.; Taylor, H. Elucidation of Fecal Inputs into the River Tagus Catchment (Portugal) Using Source-Specific Mitochondrial DNA, HAdV, and Phage Markers. Sci. Total Environ. 2021, 783, 147086. [Google Scholar] [CrossRef]
- Berry, I.M.; Melendrez, M.C.; Bishop-Lilly, K.A.; Rutvisuttinunt, W.; Pollett, S.; Talundzic, E.; Morton, L.; Jarman, R.G. Next Generation Sequencing and Bioinformatics Methodologies for Infectious Disease Research and Public Health: Approaches, Applications, and Considerations for Development of Laboratory Capacity. J. Infect. Dis. 2019, 221, jiz286. [Google Scholar] [CrossRef]
- Amdiouni, H.; Faouzi, A.; Fariat, N.; Hassar, M.; Soukri, A.; Nourlil, J. Detection and Molecular Identification of Human Adenoviruses and Enteroviruses in Wastewater from Morocco: Molecular Identification of HAdV and EV. Lett. Appl. Microbiol. 2012, 54, 359–366. [Google Scholar] [CrossRef]
- Casas, I.; Avellon, A.; Mosquera, M.; Jabado, O.; Echevarria, J.E.; Campos, R.H.; Rewers, M.; Perez-Breña, P.; Lipkin, W.I.; Palacios, G. Molecular Identification of Adenoviruses in Clinical Samples by Analyzing a Partial Hexon Genomic Region. J. Clin. Microbiol. 2005, 43, 6176–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogorzaly, L.; Walczak, C.; Galloux, M.; Etienne, S.; Gassilloud, B.; Cauchie, H.-M. Human Adenovirus Diversity in Water Samples Using a Next-Generation Amplicon Sequencing Approach. Food Environ. Virol. 2015, 7, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.-T.; Phanikumar, M.S.; Xagoraraki, I.; Rose, J.B. Quantitative Detection of Human Adenoviruses in Wastewater and Combined Sewer Overflows Influencing a Michigan River. Appl. Environ. Microbiol. 2010, 76, 715–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, V.; Chen, J.; Hsu, B.; Hsu, G.; Wang, J.; Hussain, B. Prevalence, Distribution, and Genotypes of Adenovirus and Norovirus in the Puzi River and Its Tributaries and the Surrounding Areas in Taiwan. GeoHealth 2021, 5, e2021GH000465. [Google Scholar] [CrossRef] [PubMed]
- Xagoraraki, I.; Kuo, D.H.-W.; Wong, K.; Wong, M.; Rose, J.B. Occurrence of Human Adenoviruses at Two Recreational Beaches of the Great Lakes. Appl. Environ. Microbiol. 2007, 73, 7874–7881. [Google Scholar] [CrossRef] [Green Version]
- Bofill-Mas, S.; Calgua, B.; Clemente-Casares, P.; La Rosa, G.; Iaconelli, M.; Muscillo, M.; Rutjes, S.; de Roda Husman, A.M.; Grunert, A.; Gräber, I.; et al. Quantification of Human Adenoviruses in European Recreational Waters. Food Environ. Virol. 2010, 2, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.; Goonetilleke, A.; Gardner, T. Human and Bovine Adenoviruses for the Detection of Source-Specific Fecal Pollution in Coastal Waters in Australia. Water Res. 2010, 44, 4662–4673. [Google Scholar] [CrossRef] [Green Version]
- Rafie, K.; Lenman, A.; Fuchs, J.; Rajan, A.; Arnberg, N.; Carlson, L.-A. The Structure of Enteric Human Adenovirus 41—A Leading Cause of Diarrhea in Children. Sci. Adv. 2021, 7, eabe0974. [Google Scholar] [CrossRef]
- Hemmi, S.; Vidovszky, M.Z.; Ruminska, J.; Ramelli, S.; Decurtins, W.; Greber, U.F.; Harrach, B. Genomic and Phylogenetic Analyses of Murine Adenovirus 2. Virus Res. 2011, 160, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Kumakamba, C.; N’Kawa, F.; Kingebeni, P.M.; Losoma, J.A.; Lukusa, I.N.; Muyembe, F.; Mulembakani, P.; Makuwa, M.; LeBreton, M.; Gillis, A.; et al. Analysis of Adenovirus DNA Detected in Rodent Species from the Democratic Republic of the Congo Indicates Potentially Novel Adenovirus Types. New Microbes New Infect. 2020, 34, 100640. [Google Scholar] [CrossRef] [PubMed]
- Diffo, J.; Ndze, V.N.; Ntumvi, N.F.; Takuo, J.-M.; Mouiche, M.M.M.; Tamoufe, U.; Nwobegahay, J.; LeBreton, M.; Gillis, A.; Schneider, B.S.; et al. DNA of Diverse Adenoviruses Detected in Cameroonian Rodent and Shrew Species. Arch. Virol. 2019, 164, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Qiu, M.; Ke, X.; Guan, W.; Li, J.; Huo, S.; Chen, S.; Zhong, X.; Zhou, W.; Xiong, Y.; et al. Detection of Novel Adenoviruses in Fecal Specimens from Rodents and Shrews in Southern China. Virus Genes 2016, 52, 417–421. [Google Scholar] [CrossRef]
- Rajan, A.; Palm, E.; Trulsson, F.; Mundigl, S.; Becker, M.; Persson, B.D.; Frängsmyr, L.; Lenman, A. Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses. Viruses 2021, 13, 298. [Google Scholar] [CrossRef]
- Thompson, S.S.; Jackson, J.L.; Suva-Castillo, M.; Yanko, W.A.; El Jack, Z.; Kuo, J.; Chen, C.-L.; Williams, F.P.; Schnurr, D.P. Detection of Infectious Human Adenoviruses in Tertiary-Treated and Ultraviolet-Disinfected Wastewater. Water Environ. Res. 2003, 75, 163–170. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Round | Primer Name | Primer Sequence (5′–3′) | Complexity | Tm (Mean) (°C) | Amplicon (bp) |
|---|---|---|---|---|---|
| 1st | FW universal | CTRGCYGTGGGYGAYAACMG | 32 | 63.94 | 1615 |
| RV universal | GAYTGRTCRTTGGTRTTCRTT | 32 | 56.50 | ||
| 2nd | FW A + F | TAYCARCCVGARCCKCAAGT | 48 | 62.11 | 1108 |
| RV A + F | AAGTTCCAYTCRTAVGTGTA | 12 | 54.75 | ||
| FW B + E | GTRGGCGACAACMGHGTGCT | 12 | 65.69 | 702 | |
| RV B + E | AAGCCAATGTARTTGGGTCTGTT | 2 | 61.51 | ||
| FW D | TTCAAACCCTACTCGGGCAC | 1 | 61.83 | 1339 | |
| RV D | TGATGGCAAAGAACTTTTGGGGC | 1 | 63.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavadas, J.; Parreira, R.; Leonardo, I.; Barreto Crespo, M.T.; Nunes, M. Mastadenovirus Molecular Diversity in Waste and Environmental Waters from the Lisbon Metropolitan Area. Microorganisms 2022, 10, 2443. https://doi.org/10.3390/microorganisms10122443
Cavadas J, Parreira R, Leonardo I, Barreto Crespo MT, Nunes M. Mastadenovirus Molecular Diversity in Waste and Environmental Waters from the Lisbon Metropolitan Area. Microorganisms. 2022; 10(12):2443. https://doi.org/10.3390/microorganisms10122443
Chicago/Turabian StyleCavadas, Joana, Ricardo Parreira, Inês Leonardo, Maria Teresa Barreto Crespo, and Mónica Nunes. 2022. "Mastadenovirus Molecular Diversity in Waste and Environmental Waters from the Lisbon Metropolitan Area" Microorganisms 10, no. 12: 2443. https://doi.org/10.3390/microorganisms10122443