
Is There a Universal Endurance Microbiota?

Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.1.1. Boston Marathon Study
2.1.2. Chongqing Half-Marathon Study
2.1.3. Competitive Cyclist Study
2.1.4. Sample Collection, Storage and DNA Extraction
2.2. Target Genera
2.3. Microbiome Assembly (Bioinformatics Pipeline)
2.4. Diversity
2.5. Relative Abundance Comparisons
2.5.1. Normality Testing
2.5.2. Overall Microbiota Community Comparisons
2.5.3. Hypothesis-Driven Approach
2.5.4. Data Exploration
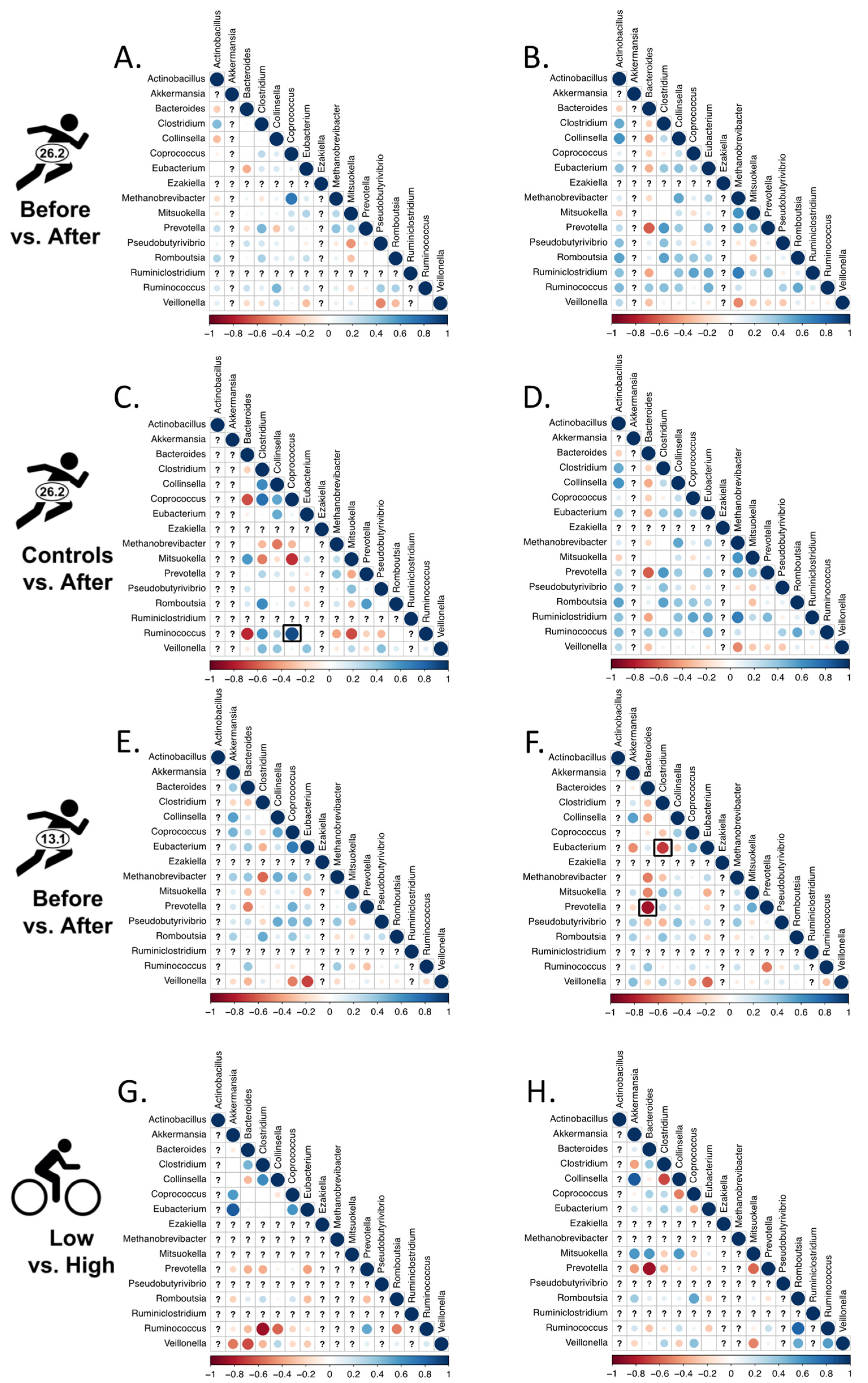
2.6. Changes in Bacterial Associations
2.6.1. Correlated Abundances and Changes Therein
2.6.2. Networks of Bacterial Associations (NetCoMi)
3. Results
3.1. Diversity
3.2. Relative Abundance
3.2.1. Normality Testing
3.2.2. Overall Microbiota Community Comparisons
3.2.3. Hypothesis-Driven Approach
3.2.4. Data Exploration (All Pairwise Comparisons)
3.3. Changes in Bacterial Associations
3.3.1. Correlated Abundances
3.3.2. Networks of Bacterial Associations (NetCoMi)
4. Discussion
4.1. Diversity
4.2. Differences in Relative Abundance
4.3. Changes in Associations
4.4. Limitations
4.5. Future Research
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lippi, G.; Favaloro, E.J.; Guidi, G.C. The Genetic Basis of Human Athletic Performance. Why Are Psychological Components so Often Overlooked? J. Physiol. 2008, 586, 3817. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Aerobic and Anaerobic Power Characteristics of Off-Road Cyclists. Med. Sci. Sports Exerc. 2001, 33, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhong, F.; Zheng, X.; Lai, H.-Y.; Wu, C.; Huang, C. Disparity of Gut Microbiota Composition Among Elite Athletes and Young Adults With Different Physical Activity Independent of Dietary Status: A Matching Study. Front. Nutr. 2022, 9, 843076. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Franzosa, E.A.; Rahnavard, G.; Hall, A.B.; Vlamakis, H.; Stevens, C.; Daly, M.J.; Xavier, R.J.; Huttenhower, C. Host Genetic Variation and Its Microbiome Interactions within the Human Microbiome Project. Genome Med. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Gérard, P.; Mosca, A.; Leclerc, M. Interplay Between Exercise and Gut Microbiome in the Context of Human Health and Performance. Front. Nutr. 2021, 8, 637010. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med. Sci. Sports Exerc. 2018, 50, 747–757. [Google Scholar] [CrossRef]
- Matsumoto, M.; Inoue, R.; Tsukahara, T.; Ushida, K.; Chiji, H.; Matsubara, N.; Hara, H. Voluntary Running Exercise Alters Microbiota Composition and Increases N-Butyrate Concentration in the Rat Cecum. Biosci. Biotechnol. Biochem. 2008, 72, 572–576. [Google Scholar] [CrossRef]
- Mitchell, C.M.; Davy, B.M.; Hulver, M.W.; Neilson, A.P.; Bennett, B.J.; Davy, K.P. Does Exercise Alter Gut Microbial Composition? A Systematic Review. Med. Sci. Sports Exerc. 2019, 51, 160–167. [Google Scholar] [CrossRef]
- Šoltys, K.; Lendvorský, L.; Hric, I.; Baranovičová, E.; Penesová, A.; Mikula, I.; Bohmer, M.; Budiš, J.; Vávrová, S.; Grones, J.; et al. Strenuous Physical Training, Physical Fitness, Body Composition and Bacteroides to Prevotella Ratio in the Gut of Elderly Athletes. Front. Physiol. 2021, 12, 670989. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Xiao, J.; Nagatomo, R.; Umemoto, H.; Morimoto, Y.; Akatsu, H.; Inoue, K.; Tsuneyama, K. Analysis of the Gut Microbiome and Plasma Short-Chain Fatty Acid Profiles in a Spontaneous Mouse Model of Metabolic Syndrome. Sci. Rep. 2017, 7, 15876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerdá, B.; Pérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; González-Soltero, R.; Larrosa, M. Gut Microbiota Modification: Another Piece in the Puzzle of the Benefits of Physical Exercise in Health? Front. Physiol. 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, R.; Mohr, A.E.; Carpenter, K.C.; Kerksick, C.M.; Purpura, M.; Moussa, A.; Townsend, J.R.; Lamprecht, M.; West, N.P.; Black, K.; et al. International Society of Sports Nutrition Position Stand: Probiotics. J. Int. Soc. Sports Nutr. 2019, 16, 62. [Google Scholar] [CrossRef] [Green Version]
- Pugh, J.N.; Sparks, A.S.; Doran, D.A.; Fleming, S.C.; Langan-Evans, C.; Kirk, B.; Fearn, R.; Morton, J.P.; Close, G.L. Four Weeks of Probiotic Supplementation Reduces GI Symptoms during a Marathon Race. Eur. J. Appl. Physiol. 2019, 119, 1491–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, E.S.; Miles, M.P.; Larson-Meyer, D.E. Dietary Supplements for Health, Adaptation, and Recovery in Athletes. Int. J. Sport Nutr. Exerc. Metab. 2018, 28, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and Associated Dietary Extremes Impact on Gut Microbial Diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, N.; Diez, G.G.; Antúnez-Almagro, C.; Bailén, M.; Bressa, C.; González Soltero, R.; Pérez, M.; Larrosa, M. A Critical Mutualism—Competition Interplay Underlies the Loss of Microbial Diversity in Sedentary Lifestyle. Front. Microbiol. 2020, 10, 3142. [Google Scholar] [CrossRef]
- Petersen, L.M.; Bautista, E.J.; Nguyen, H.; Hanson, B.M.; Chen, L.; Lek, S.H.; Sodergren, E.; Weinstock, G.M. Community Characteristics of the Gut Microbiomes of Competitive Cyclists. Microbiome 2017, 5, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keohane, D.M.; Woods, T.; O’Connor, P.; Underwood, S.; Cronin, O.; Whiston, R.; O’Sullivan, O.; Cotter, P.; Shanahan, F.; Molloy, M.G.M. Four Men in a Boat: Ultra-Endurance Exercise Alters the Gut Microbiome. J. Sci. Med. Sports 2019, 22, 1059–1064. [Google Scholar] [CrossRef]
- Kulecka, M.; Fraczek, B.; Mikula, M.; Zeber-Lubecka, N.; Karczmarski, J.; Paziewska, A.; Ambrozkiewicz, F.; Jagusztyn-Krynicka, K.; Cieszczyk, P.; Ostrowski, J. The Composition and Richness of the Gut Microbiota Differentiate the Top Polish Endurance Athletes from Sedentary Controls. Gut Microbes 2020, 11, 1374–1384. [Google Scholar] [CrossRef]
- Bressa, C.; Bailén-Andrino, M.; Pérez-Santiago, J.; González-Soltero, R.; Pérez, M.; Montalvo-Lominchar, M.G.; Maté-Muñoz, J.L.; Domínguez, R.; Moreno, D.; Larrosa, M. Differences in Gut Microbiota Profile between Women with Active Lifestyle and Sedentary Women. PLoS ONE 2017, 12, e0171352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabone, M.; Bressa, C.; García-Merino, J.A.; Moreno-Pérez, D.; Van, E.C.; Castelli, F.A.; Fenaille, F.; Larrosa, M. The Effect of Acute Moderate-Intensity Exercise on the Serum and Fecal Metabolomes and the Gut Microbiota of Cross-Country Endurance Athletes. Sci. Rep. 2021, 11, 3558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Z.; Hu, B.; Huang, W.; Yuan, C.; Zou, L. Response of Gut Microbiota to Metabolite Changes Induced by Endurance Exercise. Front. Microbiol. 2018, 9, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintikka, J.E.; Munukka, E.; Valtonen, M.; Luoto, R.; Ihalainen, J.K.; Kallonen, T.; Waris, M.; Heinonen, O.J.; Ruuskanen, O.; Pekkala, S. Gut Microbiota and Serum Metabolome in Elite Cross-Country Skiers: A Controlled Study. Metabolites 2022, 12, 335. [Google Scholar] [CrossRef]
- Lensu, S.; Pekkala, S. Gut Microbiota, Microbial Metabolites and Human Physical Performance. Metabolites 2021, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Scheiman, J.; Luber, J.M.; Chavkin, T.A.; MacDonald, T.; Tung, A.; Pham, L.-D.; Wibowo, M.C.; Wurth, R.C.; Punthambaker, S.; Tierney, B.T.; et al. Meta-Omics Analysis of Elite Athletes Identifies a Performance-Enhancing Microbe That Functions via Lactate Metabolism. Nat. Med. 2019, 25, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J.D. Diet, Microbiota, and Microbial Metabolites in Colon Cancer Risk in Rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as Biomarkers of Diet and Lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-N.; Fang, J.-Y. Gut Microbiota and Colorectal Cancer. Gastrointest. Tumors 2015, 2, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Peschel, S.; Müller, C.L.; von Mutius, E.; Boulesteix, A.-L.; Depner, M. NetCoMi: Network Construction and Comparison for Microbiome Data in R. Brief. Bioinform. 2021, 22, bbaa290. [Google Scholar] [CrossRef]
- Rintala, A.; Pietilä, S.; Munukka, E.; Eerola, E.; Pursiheimo, J.P.; Laiho, A.; Pekkala, S.; Huovinen, P. Gut microbiota analysis results are highly dependent on the 16S rRNA gene target region, whereas the impact of DNA extraction is minor. J. Biomol. Tech. 2017, 28, 19. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a Computer Program for Defining Operational Taxonomic Units and Estimating Species Richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, F.; Tadeo, R.; Voigt, A.Y.; Bork, P.; Raes, J. LotuS: An efficient and user-friendly OTU processing pipeline. Microbiome 2014, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Barco, R.A.; Garrity, G.M.; Scott, J.J.; Amend, J.P.; Nealson, K.H.; Emerson, D. A genus definition for bacteria and archaea based on a standard genome relatedness index. MBio 2020, 14, e02475-19. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network Analysis Methods for Studying Microbial Communities: A Mini Review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef]
- Badri, M.; Kurtz, Z.D.; Bonneau, R.; Müller, C.L. Shrinkage Improves Estimation of Microbial Associations under Different Normalization Methods. NAR Genom. Bioinform. 2020, 2, lqaa100. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.J.; Xu, Z.; Amir, A.; Peddada, S.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vazquez-Baeza, Y.; Birmingham, A.; et al. Effects of Library Size Variance, Sparsity, and Compositionality on the Analysis of Microbiome Data; PeerJ Inc.: Corte Madera, CA, USA, 2015. [Google Scholar]
- Strimmer, K. Fdrtool: A Versatile R Package for Estimating Local and Tail Area-Based False Discovery Rates. Bioinformatics 2008, 24, 1461–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauset, A.; Newman, M.E.J.; Moore, C. Finding Community Structure in Very Large Networks. Phys. Rev. E 2004, 70, 066111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.J. The Mathematics of Networks. In The New Palgrave Encyclopedia of Economics; Palgrave Macmillan: London, UK, 2008; pp. 1–12. [Google Scholar] [CrossRef]
- Zamkovaya, T.; Foster, J.S.; de Crécy-Lagard, V.; Conesa, A. A Network Approach to Elucidate and Prioritize Microbial Dark Matter in Microbial Communities. ISME J. 2021, 15, 228–244. [Google Scholar] [CrossRef]
- Qannari, E.M.; Courcoux, P.; Faye, P. Significance Test of the Adjusted Rand Index. Application to the Free Sorting Task. Food Qual. Prefer. 2014, 32, 93–97. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R.; Poulsen, S.K.; Larsen, T.M.; Bahl, M.I. Microbial Enterotypes, Inferred by the Prevotella-to-Bacteroides Ratio, Remained Stable during a 6-Month Randomized Controlled Diet Intervention with the New Nordic Diet. Appl. Environ. Microbiol. 2014, 80, 1142–1149. [Google Scholar] [CrossRef] [Green Version]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of Intestinal Prevotella copri Correlates with Enhanced Susceptibility to Arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The Microbiome of Professional Athletes Differs from That of More Sedentary Subjects in Composition and Particularly at the Functional Metabolic Level. Gut 2018, 67, 625–633. [Google Scholar] [CrossRef]
- Barton, W.; Cronin, O.; Garcia-Perez, I.; Whiston, R.; Holmes, E.; Woods, T.; Molloy, C.B.; Molloy, M.G.; Shanahan, F.; Cotter, P.D.; et al. The Effects of Sustained Fitness Improvement on the Gut Microbiome: A Longitudinal, Repeated Measures Case-study Approach. Transl. Sports Med. 2021, 4, 174–192. [Google Scholar] [CrossRef]
- O’Donovan, C.M.; Madigan, S.M.; Garcia-Perez, I.; Rankin, A.; O’ Sullivan, O.; Cotter, P.D. Distinct Microbiome Composition and Metabolome Exists across Subgroups of Elite Irish Athletes. J. Sci. Med. Sports 2020, 23, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Suzuki, Y. Alterations in Intestinal Microbiota in Ultramarathon Runners. Sci. Rep. 2022, 12, 6984. [Google Scholar] [CrossRef]
- Craven, J.; Cox, A.J.; Bellinger, P.; Desbrow, B.; Irwin, C.; Buchan, J.; McCartney, D.; Sabapathy, S. The Influence of Exercise Training Volume Alterations on the Gut Microbiome in Highly-Trained Middle-Distance Runners. Eur. J. Sport Sci. 2022, 22, 1222–1230. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Wegener, M.; May, S.; Schrinner, F.; Akhtar, A.; Boysen, T.J.; Schaeffer, E.; Hansen, C.; Schmidt, T.; Rühlemann, M.C.; et al. Short-Term Physical Exercise Impacts on the Human Holobiont Obtained by a Randomised Intervention Study. BMC Microbiol. 2021, 21, 162. [Google Scholar] [CrossRef]
- Aya, V.; Flórez, A.; Perez, L.; Ramírez, J.D. Association between Physical Activity and Changes in Intestinal Microbiota Composition: A Systematic Review. PLoS ONE 2021, 16, e0247039. [Google Scholar] [CrossRef]
- Realin, J.G.P.; Amoranto, M.B.C.; Parungao, M. The Gut Potential: Exploring the Modulatory Effects of Exercise and Diet on the Structure of the Gut Microbiota. Eng. J. 2021, 14, 21. [Google Scholar]
- Morishima, S.; Aoi, W.; Kawamura, A.; Kawase, T.; Takagi, T.; Naito, Y.; Tsukahara, T.; Inoue, R. Intensive, Prolonged Exercise Seemingly Causes Gut Dysbiosis in Female Endurance Runners. J. Clin. Biochem. Nutr. 2021, 68, 253–258. [Google Scholar] [CrossRef]
- Munukka, E.; Ahtiainen, J.P.; Puigbó, P.; Jalkanen, S.; Pahkala, K.; Keskitalo, A.; Kujala, U.M.; Pietilä, S.; Hollmén, M.; Elo, L.; et al. Six-Week Endurance Exercise Alters Gut Metagenome That Is Not Reflected in Systemic Metabolism in Over-Weight Women. Front. Microbiol. 2018, 9, 2323. [Google Scholar] [CrossRef] [Green Version]
- Morita, E.; Yokoyama, H.; Imai, D.; Takeda, R.; Ota, A.; Kawai, E.; Hisada, T.; Emoto, M.; Suzuki, Y.; Okazaki, K. Aerobic Exercise Training with Brisk Walking Increases Intestinal Bacteroides in Healthy Elderly Women. Nutrients 2019, 11, 868. [Google Scholar] [CrossRef] [Green Version]
- Motiani, K.K.; Collado, M.C.; Eskelinen, J.J.; Virtanen, K.A.; Löyttyniemi, E.; Salminen, S.; Nuutila, P.; Kalliokoski, K.K.; Hannukainen, J.C. Exercise Training Modulates Gut Microbiota Profile and Improves Endotoxemia. Med. Sci. Sports Exerc. 2020, 52, 94–104. [Google Scholar] [CrossRef]
- Grosicki, G.J.; Durk, R.P.; Bagley, J.R. Rapid Gut Microbiome Changes in a World-Class Ultramarathon Runner. Physiol. Rep. 2019, 7, e14313. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Tanisawa, K.; Sun, X.; Kubo, T.; Hoshino, Y.; Hosokawa, M.; Takeyama, H.; Higuchi, M. Effects of Short-Term Endurance Exercise on Gut Microbiota in Elderly Men. Physiol. Rep. 2018, 6, e13935. [Google Scholar] [CrossRef]
- Miranda-Comas, G.; Petering, R.C.; Zaman, N.; Chang, R. Implications of the Gut Microbiome in Sports. Sports Health 2022, 14, 19417381211060010. [Google Scholar] [CrossRef]
- Morueta-Holme, N.; Blonder, B.; Sandel, B.; McGill, B.J.; Peet, R.K.; Ott, J.E.; Violle, C.; Enquist, B.J.; Jørgensen, P.M.; Svenning, J.-C. A Network Approach for Inferring Species Associations from Co-Occurrence Data. Ecography 2016, 39, 1139–1150. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Lima-Mendez, G.; Lerat, J.-S.; Sathirapongsasuti, J.F.; Knight, R.; Huttenhower, C.; Lenaerts, T.; Raes, J. Cross-Biome Comparison of Microbial Association Networks. Front. Microbiol. 2015, 6, 1200. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.; Reinke, S.N.; Ali, A.; Palmer, D.J.; Christophersen, C.T. Fecal sample collection methods and time of day impact microbiome composition and short chain fatty acid concentrations. Sci. Rep. 2021, 11, 13964. [Google Scholar] [CrossRef]
- Murtaza, N.; Burke, L.M.; Vlahovich, N.; Charlesson, B.; O’ Neill, H.; Ross, M.L.; Campbell, K.L.; Krause, L.; Morrison, M. The Effects of Dietary Pattern during Intensified Training on Stool Microbiota of Elite Race Walkers. Nutrients 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Fuks, G.; Elgart, M.; Amir, A.; Zeisel, A.; Turnbaugh, P.J.; Soen, Y.; Shental, N. Combining 16S RRNA Gene Variable Regions Enables High-Resolution Microbial Community Profiling. Microbiome 2018, 6, 17. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic Data Assembly—The Way of Decoding Unknown Microorganisms. Front. Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, F.E. Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis; Springer: New York, NY, USA, 2001. [Google Scholar]
- Pugh, J.N.; Lydon, K.M.; O’Donovan, C.M.; O’Sullivan, O.; Madigan, S.M. More than a Gut Feeling: What Is the Role of the Gastrointestinal Tract in Female Athlete Health? Eur. J. Sport Sci. 2022, 22, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Jaago, M.; Timmusk, U.S.; Timmusk, T.; Palm, K. Drastic Effects on the Microbiome of a Young Rower Engaged in High-Endurance Exercise After a Month Usage of a Dietary Fiber Supplement. Front. Nutr. 2021, 8. [Google Scholar] [CrossRef]
- Jang, L.G.; Choi, G.; Kim, S.W.; Kim, B.Y.; Lee, S.; Park, H. The combination of sport and sport-specific diet is associated with characteristics of gut microbiota: An observational study. J. Int. Soc. Sports Nutr. 2019, 16, 21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event and Location | Treatment Group | Sample Size | Sampling Frequency | Reference | No. of Genera Detected |
|---|---|---|---|---|---|
| Boston Marathon, Boston, MA, USA | Runners Before | 15 | Multiple samples taken before the event | [26] | 221 |
| Boston Marathon, Boston, MA, USA | Runners After | 15 (paired with above) | Multiple samples taken after the event | [26] | 233 |
| Boston Marathon, Boston, MA, USA | Sedentary Controls | 10 | Multiple samples taken from controls | [26] | 228 |
| Chongqing International Half Marathon, Chongqing, China | Runners Before | 20 runners 1 | Once before the event | [23] | 194 |
| Chongqing International Half Marathon, Chongqing, China | Runners After | 20 (paired with above) | Once after the event | [23] | 197 |
| Competitive Cyclists, USA 2 | Low (6–10 h/wk) | 8 | One time point | [18] | 115 |
| Competitive Cyclists, USA 2 | Medium (11–15 h/wk) | 17 | One time point | [18] | 133 |
| Competitive Cyclists, USA 2 | High (16–20+ h/wk) | 8 | One time point | [18] | 115 |
| Event | Treatment Group | Number of Individuals | Number of Bacterial Genera | R-Value | p-Value | MDS Stress |
|---|---|---|---|---|---|---|
| Boston Marathon | Athletes Before vs. Athletes After | 15 (paired) | 282 | −0.021 | 0.6556 | 0.093 |
| Boston Marathon | Controls vs. Athletes After | 10 vs. 15 | 282 | 0.093 | 0.0998 | 0.074 |
| Half Marathon | Athletes Before vs. Athletes After | 19 (paired) | 198 | 0.002 | 0.3603 | 0.104 |
| Professional Cyclists | Low- vs. High-Intensity-Training Group | 8 vs. 8 | 148 | 0.517 | 0.0012 | 0.060 |
| Genera | Boston Marathon | Chongqing Half Marathon | USA Competitive Cyclists 3 | |
|---|---|---|---|---|
| “Athletes Before” vs. “Athletes After” | “Athletes After” vs. Controls | |||
| Actinobacillus | NS 1 | NS | NA | NA |
| Akkermansia | NA 2 | NA | 0.038 (+) | NS |
| Bacteroides | 0.07 | 0.09 | NS | 0.0070 (−) |
| Clostridium | 0.04 (+) | NS | NS | 0.065 |
| Collinsella | NS | NS | 0.0005 (+) | NS |
| Coprococcus | NS | NS | 0.0003 (+) | NS |
| Eubacterium | 0.07 | NS | 0.023 (+) | NS |
| Ezakiella | NA | NA | NA | NA |
| Methanobrevibacter | NS | NS | 0.09 | NA |
| Mitsuokella | NS | NS | 0.0046 (+) | NS |
| Prevotella | NS | NS | 0.07 | 0.00031 (+) |
| Pseudobutyrivibrio | NS | NS | NS | NA |
| Romboutsia | NS | NS | 0.0002 (−) | 0.0047 (+) |
| Ruminiclostridium | NS | NS | NA | NA |
| Ruminococcus | NS | NS | 0.001 (+) | NS |
| Veillonella | NS | 0.0019 (+) | 0.0004 (+) | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olbricht, H.; Twadell, K.; Sandel, B.; Stephens, C.; Whittall, J.B. Is There a Universal Endurance Microbiota? Microorganisms 2022, 10, 2213. https://doi.org/10.3390/microorganisms10112213
Olbricht H, Twadell K, Sandel B, Stephens C, Whittall JB. Is There a Universal Endurance Microbiota? Microorganisms. 2022; 10(11):2213. https://doi.org/10.3390/microorganisms10112213
Chicago/Turabian StyleOlbricht, Hope, Kaitlyn Twadell, Brody Sandel, Craig Stephens, and Justen B. Whittall. 2022. "Is There a Universal Endurance Microbiota?" Microorganisms 10, no. 11: 2213. https://doi.org/10.3390/microorganisms10112213